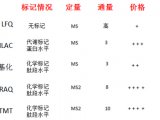
近一段时间,一直不断有Mann的新技术出现,THERMO的仪器基本上都是Mann首先发出的文章;而Aebersold自从回到瑞士以后在技术方面出的东西不多,今年ASMS质谱年会发布了和AB共同研究的SWATH技术。因未参会,一直也没有搞清是什么技术,这次在日内瓦的HUPO会议上,有幸听Aebersold本人讲了一下,对这个技术有了一个全面的了解。个人认为:这个技术有可能是蛋白质组学技术发展的一个里程碑,有取代目前DDA(data-dependent acquisition)技术的可能性。

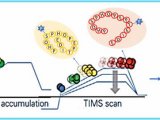
瑞士日内瓦Matthias_MannRuedi Albersold SWATH技术采用对全质量范围(
400 - 2,000 Da)进行分段做MS/MS的方式,应用AB公司的5600质谱仪,
可以在1秒内完成全质量范围的分析,所得数据应用MRMAtlas数据库进行比对分析,可以得到
低到100个拷贝的蛋白质的鉴定。这种方式不仅灵敏度高,而且通量远超过目前的DDA技术。
以前也有类似的技术,如WATERS公司的MS
E和Mann团队不久前开发的AIF技术,这些技术都是对
全谱进行MS/MS分析,虽然也可以提高鉴定效率,但因为全谱复杂度很高,增加了数据分析难度。
SWATH把全质量范围分段,借鉴了以前的GPF技术,降低了谱图的复杂性,增加了动态范围,同时应用现有数据库比对的方式进行数据分析,解决了谱图解析的难题。而5600质谱仪的高扫描速度和高质量精度为此技术的实现提供了仪器方面的保证。
可以肯定的是,这个新技术会对目前的DDA分析技术产生巨大的冲击!不过,因为此技术的主要难点是在软件方面,随着THERMO推出Q-EXACTIVE,不知是否THERMO也会来抢占这个技术要点,希望到时可以给用户更多的选择机会。
TripleTOF™ 5600质谱系统
SWATH-MS: A NEW DATA INDEPENDENT ACQUISITION LC-MS METHODOLOGY FOR QUANTITATIVE COMPLETE PROTEOME ANALYSIS
P. Navarro (1) , L. Gillet (1) , C. Carapito (2) , H. Röst (1) , L. Reiter (3) , O. Rinner (3) , S. Tate (4) , R. Bonner (4) , L. Malmström (1) , R. Aebersold (1) .
(1) Institute of Molecular Systems Biology, ETH Zürich, (2) CNRS, Université de Strasbourg, (3) Biognosys AG, (4) ABSciex.
The analysis of biomolecules in complex sample mixtures has relied for many years on LC-MS. In proteomics selected reaction monitoring (SRM) is a particularly attractive technique in cases in which reproducible data sets with high quantitative accuracy and wide dynamic range are required [1,2]. Despite the advances of SRM, the method presents certain limitations: the method requires a preliminary selection of reactions, and allows monitoring of a limited number of analytes per run. 因其重现性好、定量高准确度、宽动态范围,SRM在蛋白质组定量中成为非常有吸引力的技术。然而SRM的局限性是:必须是目标物,一次分析的数量有限。
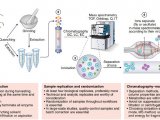
To overcome these limitations we introduce a new strategy to acquire fragment ion spectra on all the analytes in a sample, by cycling a sequence of precursor ion selection windows in the mass analyzer that collectively cover the whole targeted mass range during the entire chromatography. These windows may be seen as an analogy of the swath acquisitions in Earth satellite scans. The collected fragment ion spectra are recorded to generate a map with the dimensions
retention time - fragment ion m/z - and intensity, for each precursor ion selection window. The data analysis is then performed in the translation of the product ion spectra acquired for each isolation window into separate LC-MS2 maps, from where the fragments, derived from a spectral library and defining any precursor of interest can be extracted and analyzed to unambiguously detect and quantify the targeted analytes in the injected sample. The confidence in the peptide identification is scored based on the mass accuracy and the relative intensities of the acquired product ion fragments compared to that of the reference spectrum and on the co-elution of the extracted ion chromatograms of these fragments. Altogether, this methodology is expected outperform former LC-MS methods in terms of identification rates, quantification speed and accuracy, reproducibility of data collection, and should therefore be of large interest for performing LC-MS analyses of samples of high complexity.
[1] Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Mol Syst Biol. 2008, 4, 222.
[2] Picotti, P.; Bodenmiller, B.; Mueller, L. N.; Domon, B.; Aebersold, R. Cell. 2009, 138, 795-806.














































 首页
首页


