2024-04-26 11:08
本篇文章已经开通留言功能,
欢迎至文末留言交流讨论!
反相色谱肽分离色谱柱和流动相研究
第四部分 表征流动相的选择性

本文对多种流动相组合进行了研究,对具有不同功能的肽探针表现出不同色谱选择性(即称为表征)的亚类之间的流动相组成进行了区分,从而揭示了多模式保留机制的可能性。由于在给定流动相条件下肽保留机制的复杂性,没有尝试解释这些,而是简单地将流动相分类为不同的组,目的是确定那些产生不同选择性的组,以用于分析肽混合物的方法开发路线图的策略。在Ascentis Express C18色谱柱用不同 pH 值(范围 1.8 – 7.8)、盐类型、离子强度、离子对试剂和离液盐/趋液盐添加剂的 51 种 RPC 流动相组成研究9 种合成探针肽(牛 GLP-2 片段)色谱选择性的差异。用主成分分析(PCA)的化学计量学工具可视化所评估的各种流动相产生的选择性差异结果。利用PCA对所评估的各种流动相参数的相对重要性进行排序。这种方法的概念在含有合成相关杂质的牛GLP-2(1-15)样品的分析中得到证实。比较C18色谱柱另外三种有明显选择性差异的固定相(烷基酰胺、氟苯基和联苯)。除离子对试剂外,C18、氟苯基和联苯类色谱柱也获得了类似的趋势,这暗示了这些发现适用于绝大多数适用于肽分析的RPC色谱柱。本研究结果强调了筛选不同pH值、离子对试剂和离子强度的多种流动相的重要性,以便最大限度地提高在复杂混合物中分离所有目标肽的可能性。
01介绍
已有大量已发表的信息关于离子对试剂[1-6]、pH值[7-10]、温度[6,7,11-13]、流动相组成[7,8,10]和不同固定相[6,7]对反相LC中肽保留和选择性预测的机理效应。Hodges、Hearn、Krokhin和Gilar团队进行了广泛的研究,以增强目前对RP色谱柱上肽保留机制的理解[6,8,9,14-26]。在C18色谱柱上,通常使用流动相pH值为2和7进行肽分离,使用或不使用离子对试剂,离子对试剂与肽分子内的质子化氨基官能团(例如:组氨酸、赖氨酸和精氨酸残基以及N端氨基)相互作用。已有报道,评估具有不同疏水性的离子对试剂对特定肽分离的潜在价值[1,5]。由于多肽在不同的有机/水性环境中可以表现出不同的二级结构,以及肽在不同pH值下表现出的净电荷和电荷分布,因此优化复杂肽混合物的分离条件并非易事。这可能导致保留机制的随着流动相组成的变化而改变。
迄今为止,尚未有一项全面的研究旨在比较和表征(即分类)由不同盐、离子对试剂和具有不同固定相的pH值组成的各种流动相。由于近年来对生物制药开发的兴趣日益浓厚,因此需要识别并选择流动相和固定相,从而为色谱工作者提供在复杂混合物中分离相关肽组分的最佳方案。本文的主要重点是利用化学计量学工具,根据流动相产生的选择性特征,将51种新型和常用的流动相组合识别和表征为不同的组合。
为分离、鉴定和定量相关肽中的杂质,必须同时实现良好的选择性和峰形。因此,除了主要关注选择性外,还需评估流动相添加剂对肽峰形的影响,以及对所选目标流动相的分析物过载的影响。文献表明,选择性和峰形都高度依赖于分析物,因此色谱工作者可以选择研究一系列不同的流动相,寻找到为特定应用产生最佳分辨率的条件非常重要。
本文是系列文章中的第四篇,该系列论文涉及使用反相色谱法(RPC)最大限度地提高肽分离的色谱选择性。该系列的第一至第三篇论文分别重点介绍了使用26个专门设计的肽探针[27]开发色谱柱表征方案、优化方案[28]的稳健性以及38个不同固定相的表征[29]。该表征方案基于33个氨基酸肽-牛GLP-2(参见表1和参考文献[27–29])确定9个选定探针之间的保留时间差异,该方案已被证明可以通过肽探针的疏水性、静电性、氢键和与固定相的芳香相互作用成功地区分不同类型的反相固定相。此外,固定相分离非对映异构体或异构体探针的能力[27]。第四篇论文将表征方案的使用扩展到51种新型和常用的MS兼容和非兼容流动相。除了不同的pH缓冲液外,评估还包括一系列疏水性和电荷不同的离子对试剂(七氟丁酸(HFBA)、三氟乙酸(TFA)、二氟乙酸(DFA)、丁基磺酸钠(BuSO3Na)和三乙胺(TEA))、离液盐高氯酸钠(NaClO4)和六氟磷酸铵(NH4PF6)或趋液盐硫酸钠或硫酸铵(Na2SO4或(NH4)2SO4), 离子强度 (IS) 和一系列杂项改性剂(甲酸 (FA)、磷酸 (H3PO4)、甲磺酸 (MSA)、甲酸铵 (NH4FA)、乙酸铵 (NH4AA) 和碳酸氢铵 (NH4HCO3))的影响(见表 2)。还评估了MS兼容添加剂在电喷雾电离质谱(ESI-MS)正电模式中产生的信号强度。用主成分分析(PCA)的高度图形化化学计量工具提供流动相添加剂之间选择性差异和相似性的简单可视化,并根据流动相成分对肽探针产生的选择性分布来识别和分组不同的亚类。
本研究将有助于选择有限数量的可用于最大限度地提高给定色谱柱的选择性的流动相。首先在具有代表性的新一代C18色谱柱上研究大范围筛选流动相。然后确定是否可以使用有限数量的流动相(显示在C18相上具有较大的选择性差异),再将这些发现外推到一系列其他不同的反相色谱柱(显示在两个流动相中提供最大的选择性[29])。希望这些发现可用于在RP-LC方法开发策略中确定合适的初始流动相和固定相组合,从而提供最佳的肽分离。
02实验
2.1. 化学品和试剂
使用的所有水、乙腈和流动相添加剂(甲酸铵 (AF) 和甲酸 (FA))均为 LC-MS 级,由 Sigma Aldrich(Poole, UK)提供。二甲基亚砜(DMSO)由Fisher Scientific(Hemel Hempstead, UK)提供。由Apigenex(Prague, Czech Republic)提供的肽全部溶解在DMSO /H2O(80:20 v/v)中,浓度为0.25 mg/mL。每种肽的碱基序列都位于[27],肽探针的进一步描述见表1。
表1
2.2. 仪器
在岛津Nexera X2 UHPLC系统(Duisburg, Germany)进行LC分离,该系统配备了两个二元泵(LC-30AD)和比例阀、脱气机(DGU-20ASR)、具有冷却功能的自动进样器(SIL-30AC)、Prominence柱式烘箱(CTO-20AC)、二极管阵列检测器(SPD-M30A)和通信总线模块(CBM-20A)。岛津单四极杆质谱仪(LCMS 2020)被用作二级检测器。LC仪器的梯度滞留体积为342 μL,系统保留体积为14 μL [32] .MS研究是在配备PDA和Waters Synapt G2-Si Q-TOF(Wilmslow, UK)的Waters Acquity I-Class上进行的。
2.3. 固定相
在同一批次的Ascentis Express C18(150×2.1 mm色谱柱尺寸,2.7 μm粒径,Supelco,Bellefonte,PA,USA)上评估了所有流动相。每个离子对都使用专用色谱柱,以避免流动相之间的记忆效应。选择Polaris Amide C18(150× 2.0 mm,3 μm,Agilent Technologies,Santa Clara,CA,USA)、Acquity CSH Fluoro Phenyl(150 × 2.1 mm,1.7 μm,Waters,Milford,MA,USA)和Ascentis Express Biphenyl(150× 2.1 mm,2.7 μm)作为参比的固定相,以确定Ascentis Express C18结果在其他类型的固定相上的适用性[29]。在Acquity CSH C18(150× 1.0 mm,1.7 μm)上进行MS响应比较,该色谱柱利用了150 × 2.1 mm色谱柱上的平移梯度。每个固定相的简要描述可以在参考文献[29]的表2中找到。水峰被用作死时间标记[32]。
2.4. 流动相表征方案
如表2所述制备预混流动相,用于A相。使用MeCN / H2O(80:20 v / v)制备B相。B相不和A相做添加剂匹配,以防止溶剂浪费,并将溶剂和实验的数量保持在可接受的水平。梯度标准化如下:40 分钟内 5.6% 至 62.5%B,在梯度顶部等度保持 2 分钟,然后在 0.1 分钟和 10 分钟重新平衡(相当于 10 个色谱柱体积)后恢复到原始条件。柱温为40°C,流速为0.3 mL/min,检测波长为215 nm,带宽8 nm,参考波长为360 nm,带宽为100 nm。在PDA之后安装了带电喷雾电离功能的岛津2020单四极杆仪器,以在适用的情况下在正电SIM模式下识别峰值。使用正ESI模式电离对Synapt G2-Si MS进行了比较不同缓冲液性能的MS研究,源温度为120°C、毛细管电压为3.5 kV、脱溶剂化温度为250°C、脱溶剂化气体流量为750 L/hr、雾化器气体压力为6.0 bar、锥体气体流量为50 L/hr和扫描时间为0.250 s,。将质量范围设置为100 – 2000以观察任何加合物的形成,并应用高分辨率模式。

2.5.软件和计算
使用LabSolutions控制液相色谱仪器并进行数据处理 (Version 5.86, Shimadzu, Duisburg, Germany). 使用(SIMCA (Version 14.1, Umetrics, Umeå, Sweden)和 Origin (Version OriginPro 2016, OriginLab, Northampton, MA, USA) 做主成分分析。使用GPMAW software (Version 9.51, Lighthouse Data, Odense, Denmark)计算电荷。
03
结果和讨论
3.1. 流动相的选择
评估由不同盐、离子强度、阴离子/阳离子对试剂、离液/趋液盐在不同pH值(pH 1.8 - 7.8)下的一系列流动相,以评估它们对一系列具有不同物理/化学性质的肽的选择性。表2包含缓冲液、pH值、总离子强度、流动相组成及其MS相容性。选择流动相组合物\\的理由可在补充材料中找到。
3.2. pH的影响
肽在疏水性RP固定相上的保留取决于流动相pH值,因为流动相pH值会影响其净电离状态(例如,天冬氨酸、谷氨酸、酪氨酸、组氨酸、赖氨酸和精氨酸残基中肽的C端和N端和/或侧链的电离),这反过来又决定了它们的亲水性以及它们与离子对试剂相互作用的倾向。表1显示了所用每种肽探针的净电荷和电离碱性/酸性部分的数量。此外,pH值会影响硅醇基团在二氧化硅基固定相上的电离态以及制造商赋予的任何氨基官能团(即带电表面杂化相),因此这将影响带电肽和电离固定相表面的静电吸引/排斥。
使用PCA评估结果,前两个主成分描述了数据集中约92%的变异性。第三个主要组成部分并没有大幅增加这一数值,反而增加了评价的复杂性。因此,该研究未使用它。第一个主成分描述了大约62%的数据变异性,并且正如预期的那样,包含与静电相互作用相关的Δ(9,1)和Δ(26,13)(即变量)[29],它们在图1中彼此截然相反。除了肽电荷对选择性的预期影响外,第一主成分还受到芳香族、酚类基团和蛋氨酸残基的不同氧化态的影响,分别由Delta值Δ(16,13)、Δ(24,13)、Δ(8a,1)表示——这些Delta值可能描述了亲水性的增加(即疏水性苯基的损失, 酚基的添加和硫化物的转化形成氢键的偶极相互作用的亚砜)。第二个主成分描述了大约 30% 的变异性,其中结果主要受基于二图中 delta 值 Δ (14,13) 和 Δ 15,13) 位置的空间参数的影响(图 1)。
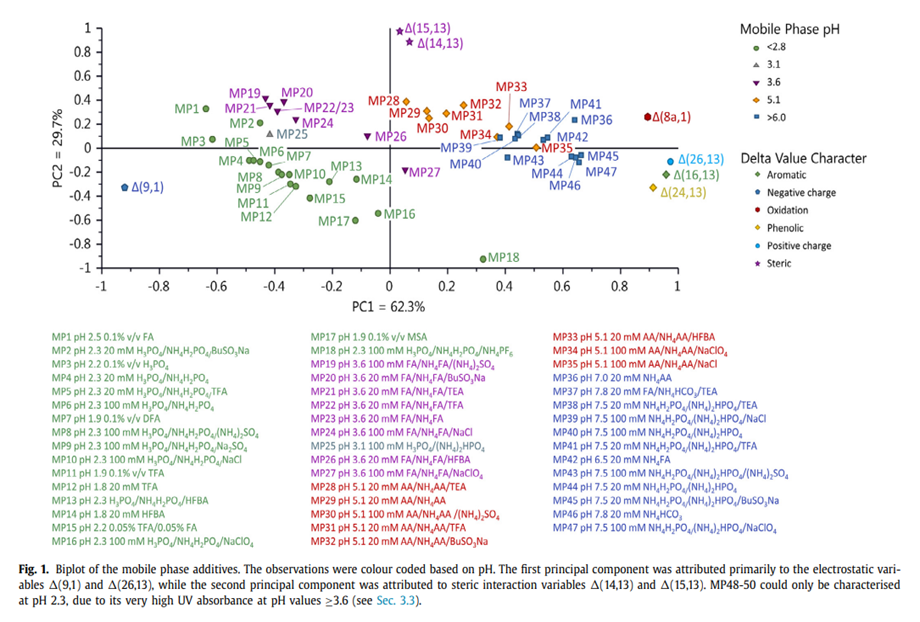
图1

图2
PCA贡献图表示两种流动相(即观测值)所赋予的选择性的差异。仅pH值变化的流动相(MP46、44、36、42、29、23和4)的贡献图(数据未显示)表明,Δ(26,13)值随pH值的增加而增加,突出了带正电荷的肽26号和带负电荷的二氧化硅表面之间的静电吸引力增加。同时,Δ (9,1) 值随着 pH 值的增加而降低,突出了带负电荷的肽 9 和带负电荷的二氧化硅表面之间的静电排斥力增加。随着流动相pH值的碱性增加(从正净电荷切换到负净电荷),1号和9号肽的保留降低,而26号肽的保留增加,即使在pH>7时,仍保留总净正电荷。在低pH值下,亲水肽的洗脱顺序为1号,在9号之前(均显示净正电荷),而在pH>7时,有一个洗脱开关,其中9号肽在1号肽之前洗脱,因为9号肽现在具有-5电荷,而1号肽(-4净电荷)因此9号肽的亲水性更强,并且对电离的硅醇基团也表现出更大的静电排斥力。有趣的是,Δ (16,13)、Δ (24,13)、Δ (8a,1) 值都随着 pH 值的增加而增加,表明随着肽羧酸部分逐渐去质子化,这些亲水项的优势增强。空间参数 Δ (14,13) 在 pH 值 3.6 和 5.1 之间最大,表明评估一系列 pH 值的重要性。图2A突出显示了在低pH值下观察到各种添加剂之间的最大选择性差异这一事实。随着流动相pH值的逐渐增加,添加剂之间观察到的选择性差异减小(即在pH 6.0时观察到流动相的扩散更大)。这可以通过在中等pH值下降低离子对形成的倾向来合理化,因为除了26号肽外,所有肽都具有负或中性净电荷。虽然疏水相互作用通常在肽的 RPC 中占主导地位,但其他类型的相互作用(如偶极子:偶极子和 π:π 相互作用)对于产生微小的保留差异可能很重要,从而可以提高选择性。结果表明,在优化肽分离的选择性时,pH值应是一个需要探索的主要参数。
使用pH 2.3、3.6、5.1和7.5的20 mM缓冲液比较含有合成杂质的牛GLP-2(1-15)样品。补充材料中的色谱图S1说明了在方法开发策略中筛选不同pH流动相时可以获得的选择性的巨大差异。由于练习的目的是说明选择性差异,因此没有做峰匹配; 而且鉴定需要2D-LC/MS,因为流动相含有不挥发性盐。
3.3. 离子对试剂的作用
先前有文章介绍,不同疏水性的肽需要不同的离子对试剂才能实现最佳分离[1,5],例如疏水性离子对试剂TFA和HFBA以及阴离子离液盐ClO4- 对亲水性肽的分离效果更好[1,3,5]。HFBA是一种非常有效的离子对试剂,可增强亲水性肽在C18色谱柱上的保留。而亲水性磷酸盐离子对试剂已被证明对疏水肽分离是成功的[1,5]。这凸显了评估具有不同表观疏水性的各种离子对试剂/反离子在分析特定肽分离方面的价值。因为在现实生活中可能存在一系列具有各种带电状态的肽,因此在 pH 值范围 1.8 至 7.8 范围内评估阴离子和阳离子对试剂的效果。
比较不同疏水性的阴离子(TFA、HFBA和BuSO3Na)和阳离子(TEA)离子对试剂对肽选择性的影响,以及没有任何电离对试剂作为流动相pH值的函数。离液添加剂NaClO4和NH4PF6也包括在内,因为它们也具有离子对特性[33]。从pH值为2.3的PCA Bi-plot图(图1)在20 mM磷酸铵缓冲液中发现,对于这种特异性分离,含有或不含TFA的流动相(MP5和4)之间的选择性差异似乎很小。与TFA相比,含有BuSO3Na和HFBA(分别为MP2和13)的流动相具有不同的选择性,这凸显了筛选不同离子对试剂的重要性。含有疏水性离子对试剂的流动相对所有携带正净电荷的肽的保留时间更长(即在pH 2.3时为+1.1至+3.4),从而产生更疏水的离子对,与C18固定相的相互作用更强。肽的保留与离子对试剂的疏水性一致(即无离子对<TFA < BuSO3Na < HFBA)。HFBA与不含离子对的流动相的暴露效果在含有几种合成杂质的牛GLP-2样品(1-15)上得到了证明(数据未显示)。
将广泛使用的0.1% v/v TFA (MP11)流动相条件与含有0.1% v/v FA (MP1)、0.1% v/v H3PO4 (MP3)和20 mM HFBA (MP14)的流动相条件进行比较,发现选择性差异显著。由于 TFA 在 质谱中产生较低的正离子 ESI 信号,因此一般将 TFA 与 FA (MP15) 结合使用或用 DFA (MP7) 代替。从PCA双图中可以看出,与TFA相比,这些替代方法之间存在小到中等的选择性差异。
一般来说,带正电荷的肽的保留随着离子对试剂的疏水性、肽上正电荷的数量和离子对试剂的浓度而增加。增加的幅度取决于肽的疏水性,例如,更多的亲水性肽比其相应的疏水类似物表现出更大的保留时间偏移[14]。这凸显了在带正电荷的肽分离优化时微调这些疏水阴离子对试剂的疏水性和/或浓度的重要性。
图 1 突出显示,离液添加剂 NaClO4 (MP16) 和 NH4PF6 (MP18) 也可以作为阴离子离子对添加剂,与其他评估的离子对试剂相比,产生的选择性特征明显不同。Hodges等人先前观察到,阴离子在形成离子对方面的有效性遵循CCl−<<TFA−<ClO4−[3]和PO4−<TFA−<HFBA−[5]的趋势,这反映了与肽形成的离子对的保留。氯化物阴离子具有亲水性,可以简单地中和肽上的正电荷,从而降低肽的整体亲水性,从而增加对RP固定相的保留。在100 mM磷酸盐中pH值为2.3的结果表明,本研究中肽的保留遵循洗脱顺序SO42− < PO4− ≈ Cl−<< ClO4− < PF6−,这是Hofmeister 级数序列的重现,Hofmeister 级数序列反应了离子影响水结构的能力 [34-35]。Hodges等人认为,ClO4−阴离子在增加肽的疏水性方面比TFA更有效,因为ClO4-阴离子具有很强的离液特性(即水结构破坏),ClO4−阴离子对附近水分子的竞争效率不如大块水团,因此比Cl−和TFA−等离子更容易脱水[3]。有人认为,离子对的形成需要将水分子排除在带正电荷和带负电荷的物质之间的相互作用之外(即阴离子必须脱水才能形成具有肽质子化氨基官能团的离子对)[3]。Hodges等人指出,ClO4−阴离子比TFA−阴离子更容易脱水,这可能部分解释了ClO4−阴离子作为离子对试剂的有效性,尽管后者更疏水[3]。
推测TFA中和了与肽相关的正电荷,并且随着其疏水性的增加,增强了肽的疏水性。
NH4PF6 (MP18) 最近在碱性小分子分析物分析中显示出良好的应用前景 [33],其在评估的阴离子离子对试剂中产生了最大的选择性差异,与 NaClO4 相比,对 +3.4 带电荷的肽编号 26 的保留率更高。有趣的是,NH4PF6 只能在 pH 值 2.3 下表征,因为它在 pH 值 ≥3.6 (MP48-50) 下的紫外吸光度非常高,因此除非在低 pH 值下使用,否则在色谱上是不切实际的。正如预期的那样,阴离子对试剂TFA、NaClO4和BuSO3Na在pH 7.5下表现出与PCA biplot图(图1)相似的选择性,因为离子对的形成比低pH值时少。在酸性环境中,所有肽探针的正电荷量明显大于负电荷,这促进了与阴离子离子对试剂的离子对形成,而在中间pH值下,肽上存在正电荷和负电荷的混合物。这产生了一个整体中性或带负电荷的肽表面,该表面不太可能与带负电荷的离子对试剂相互作用。此外,肽上的任何负电荷都可能排斥阴离子对试剂,从而减少离子对的形成[36]。在pH 6.8下含有疏水性HFBA阴离子对试剂的MP51强调了这一点,因为亲水肽(肽编号1、8和9,净电荷为-4至-5)由于与吸附在C18表面的HFBA阴离子的静电排斥而无保留。在控制保留方面,肽上带电官能团的位置及其可及性和形成离子对或与其他离子物质相互作用的能力可能与肽的总净电荷一样重要(见表1)。据报道,HFBA是一种用于分离肽的可行离子对试剂[17,30,37-39],在pH值在2.3至5.1之间,与不含任何离子对试剂的流动相相比,HFBA具有不同的选择性,如它们在评分图中的位置所示(图2B)。这已通过含有合成杂质的牛GLP-2(1-15)样品进行了验证(数据未显示)。HFBA的使用不如TFA,因为已知HFBA在LC系统中会引起记忆效应和UV基线噪音,因此需要对LC/MS仪器进行大量清洁[40]。记忆效应[41]是由于这些疏水离子对对疏水固定相具有非常强的亲和力。原则上,它们可以通过用有机溶剂洗涤来去除,但是,在实践中,色谱柱的初始特性可能已经永久改变,因此色谱柱无法再生回其原始状态。通常,这就意味着一旦色谱柱暴露于离子对试剂中,它就应该专用于该特定分析。全氟添加剂也存在潜在问题,因为含有聚四氟乙烯AF(无定形含氟聚合物)的LC管路可能会随着时间的推移而受到影响,导致聚合物的物理变化和流动相的污染[42,43]。
在pH 5.1下,阳离子对试剂TEA(MP28)产生的选择性特征与阴离子离子对试剂(MP31、32和33)不同。在低pH值下,阴离子对试剂NH4PF6和BuSO3Na之间的离子对试剂之间的选择性差异最大(图2B),其中所有评估的肽在pH 2.3下具有+1.1至+3.4的总电荷。在图2B中绘制的每个观察值(即流动相)的坐标突出显示,从低pH值到中等pH值存在收敛趋势(图2B)。正如预期的那样,所有离子对试剂的行为都相似,在低pH值下选择性差异更大,由于缺乏离子对形成,在中等pH值下,由于大多数肽表现出中性或负净电荷,因此选择性差异变窄。
甲磺酸(MSA)具有很有前途的替代选择性[30,31]、紫外线透明度和MS兼容性,在RPC作为小分子和大生物分子(如单克隆抗体)的添加剂的使用有所增加。McCalley认为,与TFA和铵盐相比,MSA可以提供不同的选择性[30]。MSA表现出最小的离子对效应,并且可能比TFA更具优势。潜在问题是存在对液相色谱体系内的金属部件腐蚀[44-46],因此强烈建议在使用后冲洗以防止部件损坏[45]。图1突出显示,与表征中使用的肽相比,MSA(MP17)产生的选择性特征不同。有趣的是,MSA导致肽的保留增强,保留顺序如下:FA<<TFA<<MSA<HFBA,表明可能与MSA形成离子对。
比较阳离子或阴离子离子对试剂对峰形影响时,在pH值范围1.8至7.8的pH范围内,超载样品获得的不对称值似乎没有差异。在没有离子对试剂的情况下,观察到较大的不对称值,特别是在中间pH值下,尤为明显。
3.4. 趋液盐和离液盐的效用
趋液性(例如SO42−离子)和离液性(例如PF6-和ClO4−)盐分别是对水结构的形成和破坏,它们会影响肽和蛋白质的溶剂化外壳,会影响肽与固定相相互作用的方式。趋液性和离液性盐都遵循Hofmeister级数[11,12],该级数描述了盐化蛋白质所需的最低浓度[47-52]。如第3.3节所示,离液盐PF6和ClO4也是阴离子对试剂,在低pH值下产生了有趣的选择性。离液剂(ClO4- 和 PF6- (分别为 MP16 和 18))在评分图中的位置(图 1 和图 2C)表明,与趋液性 SO42− 和 Cl− 盐(即 MP9 和 10)相比,选择性差异要大得多。评估的两种趋液盐(NH4)2SO4和Na2SO4(分别为MP8和9)显示出很小的选择性差异,这表明阳离子对选择性的影响很小。一般而言,使用SO42−添加剂可获得良好的峰形,但是,使用含有SO42−的流动相,9种肽中的3种肽存在明显的拖尾现象(见图3B)。相比之下,当不添加盐或添加NaCl时,观察到出色的峰形(图3A和C,总IS为100 mM)。观察到的三个肽编号 16、24 和 26 的不良形状可归因于“盐析效应”,这与肽的总净电荷无关。

图3
ClO4- 离液盐具有明显的选择性差异,尽管出于健康、安全和环境考虑,不建议使用 ClO4- 。然而,如果关键物质难以解决,它可能是一种有用的替代流动相添加剂。PF6-添加剂在选择性方面也存在更大的差异,并且不表现出爆炸性。PF6-显然是一种有趣的添加剂,然而,目前其长期使用的经验有限,还存在PF6-在水溶液中产生HF的潜在风险,这可能导致固定相的配基断裂[53]。
3.5. 离子强度和峰形
在流动相中加入盐旨在抑制二氧化硅表面带负电荷的硅醇基团与带正电荷的肽之间的静电相互作用,从而改善峰形。使用20mM 离子强度的缓冲液评估盐的效果,通过添加盐提供的总离子强度为100mM,再进行比较。离子强度已被证明对色谱性能至关重要[15,54-57]。由于多种保留机制的混合物,色谱柱上分析物的质量过载会产生不对称/拖尾峰。对于肽为带电物质,通常会相互排斥,这种排斥随着肽浓度在固定相中的增加而增加。通过增加离子强度,相互排斥减少,如McCalley等[57]所述。
在非超载状态下用表征方案[29]对相关肽进行色谱分析,此时峰容量和不对称性的影响不大。对牛GLP-2(1-15)的超载样品进行了比较,以突出在峰形上使用增加离子强度的优势。使用100 mM 离子强度的盐和离液盐的对超载牛GLP-2(1-15)样品进行研究,其不对称值范围为0.90至2.69(平均1.40),与20 mM IS流动相(0.99至4.21,平均2.82)相比有所改善。表2中指定的流动相添加剂仅在A溶剂中制备,因此在洗脱点的柱出口中实际的离子强度较低(即30%)。还评估了常用的添加剂 0.1% v/v FA、0.1% v/v TFA 和 20 mM IS TFA(图 4)。

图4
在制药行业中,经常使用磷酸盐缓冲液代替TFA,因为峰形通常更优越,可以改善UV基线,可观察到较低的定量限。超载牛GLP-2(1-15)样品在离子强度流动相低于10 mM时表现出明显的拖尾现象,特别是0.1% v/v FA。0.1% v/v TFA和20 mM TFA的不对称性得到改善,但峰的拖尾大于2.0。在洗脱峰期间,离子强度大于20 mM的流动相通常接近高斯对称,说明该肽的色谱分析需要增加离子强度。盐的类型似乎对峰的对称性没有太大影响。这证实了Hodges等人报告的发现,他们观察到含有50 mM NaCl和NaClO4盐的流动相改善了峰形[15]。图5显示了改善的峰形,该图比较了pH 2.5 0.1% v/v FA和pH 3.6 20 mM NH4FA / FA流动相对含有合成杂质的牛GLP-2样品(1-15)峰形的影响。

与常用的磷酸铵、钠或硫酸铵(图3B)相比,它们存在有机相水平升高后带来的盐沉淀和/或肽沉淀的潜在问题,已经证明NaCl(图3C)可能是使用硫酸盐或磷酸盐的可行替代品,因为与硫酸盐相比,NaCl在100 mM离子强度下产生更好的峰形,并且比磷酸盐具有更好的溶解度。然而,在低pH值下使用NaCl时必须小心,因为它可能会转化为HCl,这可能会对LC系统产生腐蚀性并限制其适用性。
3.6. 固定相对流动相的影响
接下来在另外三种色谱上不同的固定相(Ascentis Express Biphenyl、Polaris Amide C18和Acquity CSH Fluoro Phenyl)上评估了六种不同的流动相,这些流动相在PCA bi-plot图(图1)中表现出中等到较大的选择性差异,在肽反相色谱柱表征方案中显示出较大的选择性差异[29]。六种不同的流动相在pH 1.9和7.5之间变化,离子强度不同。选择0.1% v/v FA pH 2.5 (MP1)和0.1% v/v TFA,pH 1.9 (MP11),因为它们可能是最常用的低pH值肽流动相添加剂,后者是一种适用于增强保留的离子对试剂。选择MS兼容和非兼容流动相,分别为20 mM NH4FA pH 6.5 (MP42)和NH4H2PO4 / (NH4)2HPO4 pH 7.5 (MP44),因为它们代表了常见的中间pH添加剂。选择含有离子对试剂 20 mM AA / NH4AA / BuSO3Na pH 5.1 (MP32) 的流动相,因为它表现出选择性差异。此外,还包括低pH流动相100 mM H3PO4 / NH4H2PO4 / (NH4)2SO4 pH 2.3 (MP8),因为诺和诺德的经验表明,它通常比普遍使用的TFA产生更好的峰形和选择性。
将不同流动相/固定相组合的选择性与先前确定的Ascentis Express C18进行比较,以确定它们在分数图中的相似性(即Ascentis Express C18的结果和结论是否可以应用于更大范围的固定相)。Ascentis Express Bibenzyl(参见补充材料图 S2B)和 Acquity CSH 氟苯基(参见补充材料图 S2C)在评估的流动相方面呈现出与 Ascentis Express C18(参见补充材料图 S2A)相似的模式。这表明中性、负电/极性和弱正电固定相的反应相似,因为它们在Score Plot图中产生了相似的模式。MP32(pH 5.1 20 mM AA / NH4AA / BuSO3Na)的行为不同,这种偏差可能是由于离子对试剂与附着在固定相表面的C18、联苯或丙基五氟苯配体相互作用[58-60]。这可能会改变肽与离子对试剂和固定相的相互作用方式,从而可能提供不同的选择性曲线。补充材料图S2C中MP44的Delta值(即Acquity CSH氟苯基在pH 7.5下)无法获得,因为亲水肽(肽编号1、8和9,净电荷为-4至-5)在过早洗脱,因为它们在这种低保留固定相表面上与负电荷的硅醇基团发生静电排斥。尽管有BuSO3Na流动相的结果,但令人鼓舞的观察结果是,中性、负电性/极性和弱正电性固定相的行为方式相似,这表明这些结果应该可以拓展应用到的市售色谱柱上。
Polaris Amide C18的分数图(见补充材料图S2D)中的模式具有很高的正电特性,表明这种类型色谱柱与中性的Ascentis Express C18之间没有相关性。此外,使用肽反相色谱柱表征方案[29],也观察到Polaris Amide C18相会产生较大的选择性差异,因此,可以合理地预期,与C18色谱柱相比,该固定相在流动相范围内的行为会有所不同。MP42 (pH 6.5) 与 MP1 (pH 2.5) 作为四个固定相的函数的贡献图(数据未显示)突出了 C18、联苯和氟苯基相响应之间的相似性。后三种在pH 6.5时由于二氧化硅表面的电离增加,与pH 2.5相比表现出更大的亲水性(Δ(8a,1),Δ(16,13),Δ(24,13)),正静电相互作用(Δ(26,13))和较低的负电排斥描述符(Δ(9,1))。相比之下,带正电荷的Polaris Amide C18固定相表现出较低的静电相互作用(Δ(26,13))和亲水性(Δ(8a,1),Δ(16,13),Δ(24,13))描述符,因为色谱柱上的正电荷抵消了pH 6.5时电离硅醇基团增加的负电荷。这种对固定相的有限评估表明,Ascentis Express C18的结果可以应用于由中性、负电/极性和弱正电固定相组成的色谱柱组,即大多数市售色谱柱。在固定相具有巨大选择性差异的情况下,流动相表征结果不太适用。
3.7. 质谱响应
1号肽(牛GLP-2(1-15))在正电模式电喷雾电离(ESI)中的MS信号强度被评估为补充材料表S1中所示的一系列挥发性流动相添加剂的函数。MS信号的强度范围在6E+02至2E+05之间,显示出不同流动相添加剂之间的MS信号强度差异很大。MS信号强度。对应于pH 2.5 0.1% v/v FA (MP1)和pH 7.8 20 mM FA / NH4HCO3 / TEA (MP37)的流动相分别给出了最大和最小的大量挥发性流动相的MS响应%(与FA(MP1)相比)在0-10%之间,其中包括一些常用的MS添加剂,如pH 6.5 20 mM NH4FA (MP42)、pH 7.0 20 mM NH4AA (MP36)和pH 7.8 20 mM NH4HCO3 (MP46)。与FA相比,普遍使用的pH 1.9 0.1% v/v TFA(MP11)仅产生20%的响应,这与先前的报道一致[38]。在流动相(MP15)中用FA代替50%的TFA部分纠正了ESI正离子灵敏度降低的情况[38,61]。由于纯度低且金属含量高,DFA历来避免作为LC/MS的添加剂[62],然而,由于生产工艺的改进,DFA(MP7)的质量最近有所提高,使其成为TFA(MP11)的可行替代品[63]。在pH 1.9的测试条件下,13 mM 0.1% v/v DFA (MP7) 产生了 50% 的响应(比 TFA 高 2.5 倍)。有趣的是,与FA相比,不常用的pH 1.9 0.1% v/v MSA(MP17)产生了15%的合理MS响应。对于所有添加剂,加合物的形成被忽略不计。
含有挥发性离子对试剂TEA(MP21、28、31和37)和HFBA(MP14、26和33)的流动相对该肽的MS信号响应较差,通常禁止其用于低杂质测量。HFBA还会引起记忆效应,需要对MS仪器进行大量清洁[40]。
在将这些发现外推到其他肽时必须谨慎,因为 MS 响应高度依赖于分析物和 MS 操作条件。甲酸通常被认为是生成高灵敏度阳性模式ESI-MS的“金标准”,但不幸的是,与TFA或磷酸盐缓冲液相比,甲酸的峰形较差[57]。当使用 TFA 时,观察到 MS 反应显着降低,这表明应考虑 用DFA 替换 TFA,因为DFA带来 MS 信号的减少并不那么明显。结果表明,在pH范围内具有不同选择性性质的挥发性流动相添加剂应产生与常用的0.1%TFA相当的可接受的MS响应。
04结论
本文使用PCA的化学计量学工具可视化不同流动相之间可能产生的巨大选择性差异。结果强调了筛选不同pH和离子对试剂的几种流动相的重要性,最大限度地提高在复杂混合物中分离所有目标肽的可能性。PCA表明,对于这一特定范围的肽,当离子对试剂在合适的pH值下使用时,它们会产生很大的选择性差异,从而促进离子对的形成。PCA评分图(图2)和几何距离(数据未显示)突出显示,阴离子离子对试剂HFBA、ClO4和PF6对低pH值下离子对试剂的选择性影响最大。4种不同色谱柱和6种不同流动相的PCA评分图和几何距离(图S2)表明,流动相参数对选择性的相对重要性为:pH值和色谱柱类型>pH>色谱柱类型>离子对试剂(但是,如果包括更多样化的离子对试剂,如HFBA、ClO4和PF6),那么离子对试剂的重要性就会增大)。
在分析含有合成相关杂质的牛GLP-2(1-15)样品时,流动相提供了不同选择性。具有高离子强度的流动相被证明对于产生对称峰至关重要。比较C18色谱柱和另外三种固定相(即烷基酰胺、氟苯基和联苯),这些固定相先前已被证明在有限的流动相子集上对这些肽具有较大的选择性差异。除离子对试剂BuSO3Na外,C18、氟苯基和联苯相的趋势相似,表明这些发现适用于绝大多数适用于肽分析的RPC色谱柱(即中性或弱极性)。这些结论与具有更不同性质(即含有高度正电荷)的色谱柱无关。
这项工作的结果与固定相表征研究[29]相结合,有望有助于定义RPC肽分离的方法开发策略。本文旨在为分析人员提供流动相相似性/差异性的快速简约可视化,以便进行方法开发选择,并不打算说明哪种流动相组成是最佳的,因为这将是特定肽应用所独有的,分析人员有责任发现和验证用于特定肽分离的最佳流动相添加剂。
反相色谱肽分离色谱柱和流动相研究
编译结语
看到本结语,可能您已经有所收获。无论您是否从事肽的有关物质方法开发或者只关注小分子的研究,通读或者完全读懂这四篇文章都会带来不少的帮助。这四篇文章中有非常丰富的化学结构和性质相关知识,色谱柱化学知识,统计学知识,耐用性研究策略等等。无论您获得多与少,总之,恭喜您完成了四万多字的阅读。
以下是我们ACD/Labs能够为从事肽类物质研究的色谱工作者提供的工具:
本系列文章中提到的肽反相色谱柱表征库以及小分子适用的Tanaka或者Tanaka 拓展色谱柱数据库在AutoChrom内皆可查询。

图1 Tanaka 色谱柱数据库

图2 拓展Tanaka 色谱柱数据库

图3 肽反相色谱柱表征数据库
除此之外,AutoChrom内还含有静电荷计算软件,位于Percetpa pKa模块内。

图4 Percepta pKa 静电荷计算器
Chemsketch 内含有缓冲盐计算器,可计算pH,离子强度和缓冲容量
图5 pH Calculator 界面计算缓冲体系
LC-Simulator 内可对影响因素进行单因素或者多因素建模
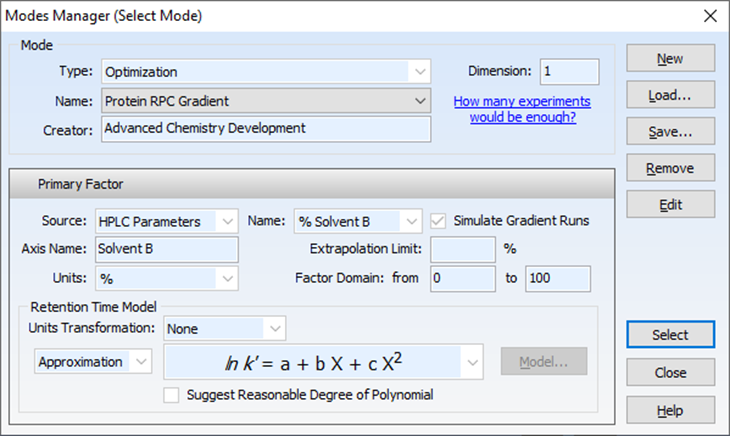
图5 蛋白质梯度建模保留时间模拟公式

图6 蛋白质梯度温度两因素建模保留时间模拟公式
Lc-Simulator三因素响应面揭示可操作范围
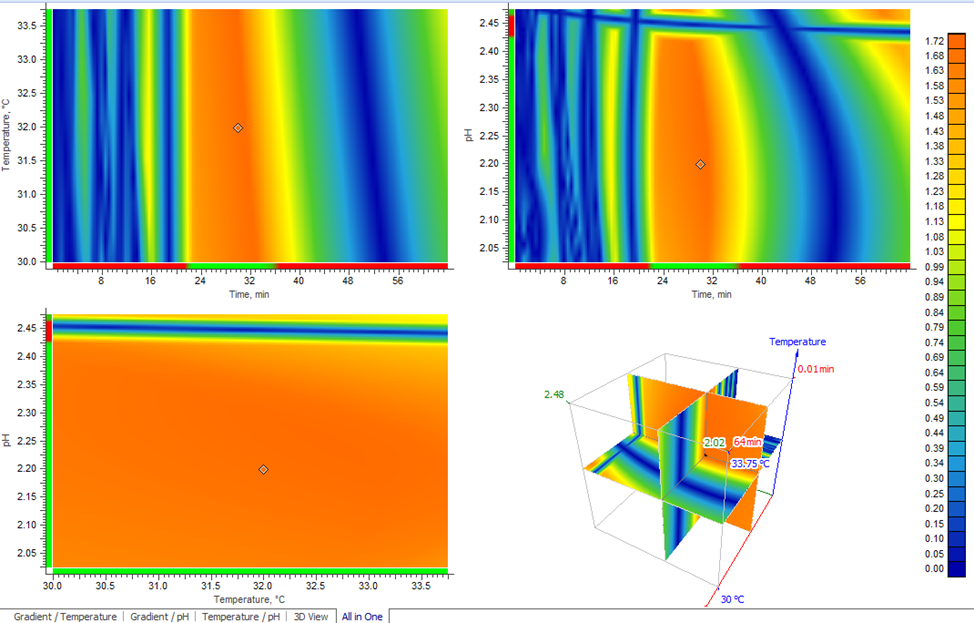
图7 三因素响应面模型展示
如果您想做更多的沟通和联络,请与我们取得联系。有很多科学工作我们可以一起探讨和协作。
请联络:
zuowei.yan@acdlabs.com
13816084932
参考文献:
向上滑动阅览
[1] D.C. Guo, C.T. Mant, R.S. Hodges, Effects of ion-pairing reagents on the prediction of peptide retention in reveresed-phase high performance liquid chromatography, J. Chromatogr. 386 (1987) 205–222.
[2] Y. Chen, A.R. Mehok, C.T. Mant, R.S. Hodges, Optimum concentration of trifluoroacetic acid for reversed-phase liquid chromatography of peptides revisited, J. Chromatogr. A. 1043 (2004) 9–18.
[3] M. Shibue, C.T. Mant, R.S. Hodges, The perchlorate anion is more effective than the trifluoroacetate anion as an ion-pairing reagent for reversed-phase chromatography of peptides, J. Chromatogr. A. 1080 (2005) 49–57.
[4] M. Shibue, C.T. Mant, R.S. Hodges, Effect of anionic ion-pairing reagent hydrophobicity on selectivity of peptide separations by reversed-phase liquid chromatography, J. Chromatogr. A. 1080 (2005) 68–75.
[5] M. Shibue, C.T. Mant, R.S. Hodges, Effect of anionic ion-pairing reagent concentration (1-60 mM) on reversed-phase liquid chromatography elution behaviour of peptides, J. Chromatogr. A. 1080 (2005) 58–67.
[6] M. Gilar, H. Xie, A. Jaworski, Utility of retention prediction model for investigation of peptide separation selectivity in reversed-phase liquid chromatography: Impact of concentration of trifluoroacetic acid, column temperature, gradient slope and type of stationary phase, Anal. Chem. 82 (2010) 265–275.
[7] Y. Chen, C.T. Mant, R.S. Hodges, Selectivity differences in the separation of amphipathic alpha-helical peptides during reversed-phase liquid chromatography at pHs 2.0 and 7.0: effects of different packings, mobile phase conditions and temperature, J. Chromatogr. A. 1043 (2004) 99–111.
[8] C.T. Mant, A. Byars, S. Ankarlo, Z. Jiang, R.S. Hodges, Separation of highly charged (+5 to +10) amphipathic α-helical peptide standards by cation-exchange and reversed-phase high performance liquid chromatography, J. Chromatogr. A. 1574 (2018) 60–70.
[9] C.T. Mant, D. Cepeniene, R.S. Hodges, Reversed-phase HPLC of peptides: Assessing column and solvent selectivity on standard polar-embedded and polar endcapped columns, J. Sep. Sci 33 (2010) 3005–3021.
[10] H. Liu, B. Xu, M.K. Ray, Z. Shahrokh, Peptide mapping with liquid chromatoraphy using a basic mobile phase, J. Chromatogr. A. 1210 (2008) 76–83.
[11] C.T. Mant, Y. Chen, R.S. Hodges, Temperature profiling of polypeptides in reversed-phase liquid chromatography I. Monitoring of dimerization and unfolding of amphipathic alpha-helical peptides, J. Chromatogr. A. 1009 (2003) 29–43.
[12] C.T. Mant, B. Tripet, R.S. Hodges, Temperature profiling of polypeptides in reversed-phase liquid chromatography II. Monitorying of folding and stability of two stranded alpha-helical coiled-coils, J. Chromatogr. A. 1009 (2003) 45–59.
[13] Y. Chen, C.T. Mant, R.S. Hodges, Temperature selectivity effects in reversed-phase liquid chromatography due to conformation differences between helical and non-helical peptides, J. Chromatogr. A. 1010 (2003) 45–61.
[14] C.T. Mant, R.S. Hodges, Context dependent effects on the hydrophilicity /hydrophobicity of side-chains during reversed-phase high performance liquid chromatography: Implications for prediction of peptide retention behaviour, J. Chromatogr. A. 1125 (2006) 211–219.
[15] J.M. Kovacs, C.T. Mant, R.S. Hodges, Determination of intrinsic hydrophilicity /hydrophobicity of amino acid side chains in peptides in the absence of nearest-neighbour or conformation effects, Biopolymers (Pept. Sci.) 84 (2006) 283–297.
[16] B. Tripet, D. Capeniene, J.M. Kovacs, C.T. Mant, O.V. Krokhin, R.S. Hodges, Requirements for prediction of peptide retention time in reversed-phase high performance liquid chromatography: Hydrophilicity /hydrophobicity of side-chains at the N- and C-termini of peptides are dramatically affected by the end groups and location, J. Chromatogr. A. 1141 (2007) 212–225.
[17] C.T. Mant, J.M. Kovacs, H.M. Kim, D.D. Pollock, R.S. Hodges, Intrinsic amino acid side chain hydrophilicity /hydrophobicity coefficients determined by reversed-phase high performance liquid chromatography of model peptide: Comparison with other hydrophilicity / hydrophobicity scales, Pept. Sci. 92 (2009) 573–595.
[18] C.T. Mant, R.S. Hodges, Design of peptide standards with the same composition and minimal sequence variation to monitor performance /selectivity of reversed-phase matrices, J. Chromatogr. A. 1230 (2012) 30–40.
[19] M. Gilar, A. Jaworski, T.S. Mcdonald, Solvent selectivity and strength in reversed-phase liquid chromatography separation of peptides, J. Chromatogr. A. (2014) 1337.
[20] M. Gilar, A. Jaworski, Retention behaviour of peptides in hydrophilic interaction chromatography, J. Chromatogr. A. 1218 (2011) 8890–8896.
[21] V. Spicer, A. Yamchuk, J. Cortens, S. Sousa, W. Ens, K.G. Standing, J.A. Wilkins, O.V. Krokhin, Sequence specific retention calculator. A family of peptide retention time prediction algorithms in reversed-phase HPLC: Applicability to various chromatographic conditions and columns, Anal. Chem. 79 (2007) 8762–8768.
[22] O.V. Krokhin, Peptide retention prediction in reversed-phase chromatography: Proteomic applications, Expert. Rev. Proteomics 9 (2012) 1–4.
[23] A.N. Hodder, M.I. Aguilar, M.T. Hearn, High performance liquid chromatography of amino acids, peptides and proteins. LXXXIX. The influence of different displacer salts on the retention properties of proteins separated by gradient anion-exchange chromatography, J. Chromatogr. 476 (1989) 391–411.
[24] A.W. Purcell, G.L. Zhao, M.I. Aguilar, M.T.W. Hearn, Comparison between the isocratic and gradient retention behaviour of polypeptides in reversed-phase liquid chromatographic environments, J. Chromatogr. A. 852 (1999) 43–57.
[25] R.I. Boysen, A.J.O. Jong, M.T.W. Hearn, Binding behaviour and conformational properties of globular proteins in the presence of immobilised non-polar ligands used in reversed-phase liquid chromatography, J. Chromatogr. A. 1079 (2005) 173–186.
[26] R.I. Boysen and M.T.W. Hearn, High-Performance Liquid Chromatography of Peptides and Proteins, in Amino Acids, Peptides and Proteins in Organic Chemistry: Analysis and Function of Amino Acids and Peptides, A.B. Hughes, Editor. 2011, Wiley: Chichester. p. 167-210.
[27] J.K. Field, M.R. Euerby, P. Petersson, J. Lau, H. Thøgersen, Investigation into reversed-phase chromatography peptide separation systems part I: Development of a protocol for column characterisation, J. Chromatogr. A. 1603 (2019) 113–129.
[28] J.K. Field, M.R. Euerby, P. Petersson, Investigation into reversed-phase chromatography peptide separation systems Part II: An evaluation of the robustness of a protocol for column characterisation, J. Chromatogr. A. 1603 (2019) 102–112.
[29] J.K. Field, M.R. Euerby, P. Petersson, Investigation into reversed phase chromatography peptide separation systems part III: Establishing a column characterisation database, J. Chromatogr. A. (2020) 1622.
[30] D.V. McCalley, Effect of mobile phase additives on solute retention at low aqueous pH in hydrophilic interaction liquid chromatography, J. Chromatogr. A., 1483 (2017) 71–79.
[31] D.V. McCalley, D. Guillarme, Evaluation of additives on reversed-phase chromatography of monoclonal antibodies using a 1000 A stationary phase, J. Chromatogr. A. (2020) 1610. [32] P. Petersson, B.O. Boateng, J.K. Field, M.R. Euerby, A practical approach to modelling of reversed-phase liquid chromatographic separations: Advantages, principles and possible pitfalls, LCGC Europe 31 (2018) 120–143.
[33] I.A. Haidar Ahmed, W. Chen, H.M. Halsey, A. Klapars, J. Limanto, G.F. Pirrone, T. Nowak, R. Bennett, R. Hartman, A.A. Makarov, I. Mangion, E.L. Regalado, Multi-column ultra-high performance liquid chromatography screening with chaotropic agents and computer-assisted separation modeling enables process development of new drug substances, Analyst 144 (2019) 2872–2880.
[34] F. Hofmeister, Instructions on the effects of mineral salts: Mechanisms of protein precipitation by mineral salts and their role in protein function, Arch. Exp. Pathol. Pharmacol. 24 (1888) 247–260.
[35] H. Jakubowski. Hofmeister Series. 2006 (Accessed 22nd December 2020)]; Available from: https://employees.csbsju.edu/hjakubowski/classes/ch331/ protstructure/hofmeister.gif.
[36] B. Lajin, W. Goessler, Fluorinated carboxylic acids as "ion repelling agents" in revered-phase chromatography, J. Chromatogr. A. (2020) 1631.
[37] D.V. McCalley, Study of retention and peak shape in hydrophilic interactions chromatography over a wide pH range, J. Chromatogr. A. 1411 (2015) 41–49.
[38] M.C. García, The effect of the mobile phase additives on sensitivity in the analysis of peptides and proteins by high-performance liquid chromatography-electrospray mass spectrometry, J. Chromatogr. B. 825 (2005) 111–123.
[39] A. Apffel, S. Fischer, G. Goldberg, P.C. Goodley, F.E. Kuhlmann, Enhanced sensitivity for peptide mapping with electrospray liquid chromatography-mass spectrometry in the presence of signal suppression due to trifluoroacetic acid containing mobile phases, J. Chromatogr. A. 712 (1995) 177–190.
[40] X. Wang, S. Yang, Y. Li, J. Zhang, Y. Jin, W. Zhao, Y. Zhang, J. Huang, P. Wang, C. Wu, J. Zhou, Optimisation and application of parallel solid-phase extraction coupled with ultra-high performance liquid chromatography-tandem mass spectrometry for the determination of 11 aminoglycoside residues in honey and royal jelly, J. Chromatogr. A. 1542 (2018) 28–36.
[41] M.C. Garcia-Alvarez-Coque, G. Ramis-Ramos, M.J. Ruiz-Angel, et al., Atomic Absorption Spectrometry - Electrothermal, in: P. Worsfold, et al. (Eds.), Encyclopedia of Analytical Science, Editors., Elsevier, 2016, pp. 117–126.
[42] , The 0.1% TFA revolution - A sign of chromatographic laziness?, in: The Column, LCGC, 13, 2017, pp. 11–13.
[43] W.H. Tuminello, G.T. Dee, Thermodynamics of poly(tetrafluoroethylene) solubility, Macromolecules 27 (1994) 669–676.
[44] B. Gaur, H.S. Srinivasan, Corrosion of metals and alloys in methane sulphonic acid, British Corrosion Journal 34 (1999) 63–68.
[45] M. Finšgar, I. Milošev, Corrosion behaviour of stainless steels in aqueous solutions of methanesulfonic acid, Corrosion Science 52 (2010) 2430–2438.
[46] M. Finšgar, J. Jackson, Electrochemical study of AISI C1018 steel in methanesulfonic acid containing an acetylenic alcohol-based corrosion inhibitor formulation, J. Lab. Automation 21 (2016) 632–641.
[47] A.M. Hyde, S.L. Zultanski, J.H. Waldman, Y. Zhong, M. Shevlin, F. Peng, General principles and strategies for salting-out informed by the Hofmeister series, Org. Process Res. Dev. 21 (2017) 1355–1370.
[48] M. Chaplin. Kosmotropes and Chaotropes. 2019 (Accessed 18th September 2019)]; Available from: http://www1.lsbu.ac.uk/water/kosmotropes_ chaotropes.html.
[49] P. Ball, J.E. Hallsworth, Water structure and chaotropicity: their uses, abuses and biological implications, Phys. Chem. Chem. Phys. 17 (2015) 8297–8305.
[50] A. Makarov, R. LoBrutto, Y. Kazakevich, Liophilic mobile phase additives in reversed phase HPLC, J. Liquid Chromatogr. & Rel. Tech. 31 (2008) 1533–1567.
[51] R. LoBrutto, A. Jones, Y.V. Kazakevich, H.M. McNair, Effect of the eluent pH and acidic modifiers in high-performance liquid chromatography retention of basic analytes, J. Chromatogr. A. 913 (2001) 173–187.
[52] J. Flieger, The effect of chaotropic mobile phase additives on the separation of selected alkaloids in reversed-phase high-performance liquid chromatography, J. Chromatogr. A. 1113 (2006) 37–44.
[53] L. Terborg, S. Nowak, S. Passerini, M. Winter, U. Karst, P.R. Haddad, P.N. Nesterenko, Ion chromatographic determination of hydrolysis products of hexafluorophosphate salts in aqeuous solution, Anal. Chim. Acta 714 (2012) 121–126.
[54] D.V. McCalley, Study of overloading of basic drugs and peptides in reversed-phase high performance liquid chromatography using pH adjustment of weak acid mobile phases suitable for mass spectrometry, J. Chromatogr. A. 1075 (2005) 57–64.
[55] D. Johnson, B. Boyes, R. Orlando, The use of ammonium formate as a mobile phase modifier for LC-MS/MS analysis of tryptic digests, J. Biomol. Tech. 24 (2013) 287–297.
[56] B. Bobály, A. Beck, J. Fekete, D. Guillarme, S. Fekete, Systematic evaluation of mobile phase additives for the LC-MS characterisation of therapeutic proteins, Talanta 136 (2015) 60–67.
[57] D.V. McCalley, Effect of buffer on peak shape of peptides in reversed-phase high performance liquid chromatography, J. Chromatogr. A. 1038 (2004) 77–84.
[58] J.W. Dolan, Ion pairing - Blessing or curse? LCGC North America 26 (2008) 170–174.
[59] A. Vailaya, C. Horvath, Retention in reversed-phase chromatography: partition or adsorption, J. Chromatogr. A. 829 (1998) 1–27.
[60] L.R. Snyder, J.J. Kirkland, J.W. Dolan, Chapter 7.4, in: Ion-Pair Chromatography (IPC), in Introduction to modern liquid chromatography, Wiley & Sons, New Jersey, 2010, pp. 331–347.
[61] A.L.L. Duchateau, B.H.M. Munsters, G.T.C. Kwakkenbos, R.G.J.V. Leuken, Selection of buffers and of an ion-pairing agent for thermospray liquid chromatographic-mass spectrometric analysis of ionic compounds, J. Chromatogr. 552 (1991) 605–612.
[62] J. Nguyen, J. Smith, O.V. Friese, J.C. Rouse, D.P. Walsh, X. Zhang, N. Ranbaduge, M.A. Lauber, A new LC-MS approach for enhancing subunit-level profiling of mABs and ADCs, ASMS, San Diego, CA, 2018.
[63] J.M. Nguyen, J. Smith, S. Rzewuski, M.A.L.C. Legido-Quigley, High sensitivity LC-MS profiling of antibody-drug conjugates with difluoroacetic acid ion pairing, MAbs 11 (2019) 1358–136
本篇文章为反相色谱肽分离系统研究第四部分,本研究共有四个部分,现已全部发布在ACD/Labs微信公众号。
前期回顾:
1. 反相色谱肽分离系统研究第一部分:开发色谱柱表征方案
2. 反相色谱肽分离系统研究 第二部分:色谱柱表征方案的耐用性评估
3. 反相色谱多肽分离系统研究第三部分:建立色谱柱表征数据库
ACD/Labs CN
微信号|ACDLabsCN