蛋白质组实验流程
双向电泳的样品的制备
样品制备原则
样品制备是双向电泳中最关键的一步,将直接影响2-DE结果好坏。目前并没有一个通用的样品制备方法,尽管处理方法多种多样,但都遵循几个基本的原则:1)尽可能的提高样品蛋白的溶解度,抽提最大量的总蛋白,减少蛋白质的损失;2)减少对蛋白质的人为修饰;3)破坏蛋白质与其他生物大分子的相互作用,并使蛋白质处于完全变性状态。
根据这一原则,样品制备需要四种主要的试剂:离液剂(chaotropes),主要包括尿素(Urea)和硫脲(thiourea);表面活性剂(sufactants),也称去垢剂,如CHAPS与Zwittergent系列等双性离子去垢剂;还原剂(reducing agents),最常用的是二硫苏糖醇(DTT)和磷酸三丁酯(TBP)等。当然,也可以选择性的加入Tris-base,蛋白酶抑制剂以及核酸酶。
样品的来源不同,其裂解的缓冲液也各不相同。通过不同试剂的合理组合,以达到对样品蛋白的最大抽提。在对样品蛋白质提取的过程中,必须考虑到去除影响蛋白质可溶性和2DE重复性的物质,比如核酸、脂、多糖等大分子以及盐类小分子。大分子的存在会阻塞凝胶孔径,盐浓度过高会降低等电聚焦的电压,甚至会损坏IPG胶条,这样都会造成2-DE的失败。样品制备的失败很难通过后续工作的完善或改进获得补偿。
核酸的去除可采用超声或核酸酶处理,超声处理应控制好条件,并防止产生泡沫;而加入的外源核酸酶则会出现在最终的2D胶上。脂类和多糖都可以通过超速离心除去。透析可以降低盐浓度,但时间太长;也可以采取凝胶过滤或沉淀/重悬法脱盐,但会造成蛋白质的部分损失。
因此,样品制备方法必须根据不同的样品、所处的状态以及实验目的和要求来进行选择。目前有很多方法适于2-DE,如组织或细胞的总蛋白提取物、亚细胞组份或细胞器蛋白、免疫沉淀的蛋白及其它亚组份蛋白(如磷酸化蛋白、采用亲合纯化凝集素结合蛋白等)。
一、细胞样品
1. 细胞培养,加药与处理。
2. 胰酶消化贴壁细胞,PBS漂洗3次(1500g,5min),弃上清, 再次离心,去尽残液(非常重要!)。如要比较细胞膜蛋白组的差别,最好用细胞刮收获细胞。如用10mM Tris/ 250 mM Sucrose(pH 7.0)代替PBS,可有效降低样品的盐浓度。
加入5倍体积裂解液,混匀(或将1×106细胞悬于60~100µl裂解液中)。
3. 加50ug/ml RNase及200ug/ml Dnase,在4℃放置15分钟。
4. 15,000转,4℃离心60分钟(或40,000转,4℃离心30分钟)。
5. 收集上清。
6. 测定蛋白浓度(采用BioRad RC/DC protein assay kit)。
7. 分装样品,冻存于-70℃。
二、组织样品
1. 碾钵碾磨组织,碾至粉末状。
2. 将适量粉末状组织转移至匀浆器,加入适量裂解液,进行匀浆。
3. 加50ug/ml RNase及200ug/ml DNase,在4℃放置15分钟。
4. 15,000转,4℃离心60分钟(或40,000转,4℃离心30分钟)。
5. 收集上清,测定蛋白浓度。
6. 分装样品,冻存于-70℃。
注意事项:
1. 8 mmol/L PMSF必须在添加还原剂之前用,否則PMSF会失去活性。
2. 40 mmol/L浓度以下的Tris可使有些蛋白酶在高pH值下失活。
3. 细胞清洗――大多用PBS,若PBS残留于细胞表面会造成胶上出现水平条纹,则可利用(10 mmol/L Tris, 250 mmol/L sucrose pH 7.0)來解決此问题。
双向电泳操作步骤
水化上样(被动上样)
1. 从冰箱中取出IPG胶条,室温放置10min。
2. 沿水化盘槽的边缘从左向右线性加入样品,槽两端各1cm左右不加样,中间的样品液一定要连贯.注意:不要产生气泡,否则会影响胶条中蛋白质的分布。
3. 用镊子轻轻撕去IPG胶条上的保护层。注意:碱性端较脆弱,应小心操作。
4. 将IPG胶条胶面朝下轻轻置于水化盘中样品溶液上.注意:不要将样品溶液弄到胶条背面,因为这些溶液不会被胶条吸收;还使胶条下面的溶液产生气泡。如产生了气泡,用镊子轻轻地提起胶条的一端,上下移动胶条,直到气泡被赶走。
5. 放置30~45min大部分样品被胶条吸收,沿着胶条缓慢加入矿物油,每根胶条约3ml(17cm IPG),防止胶条水化过程中液体蒸发。
6. 置等电聚焦仪于-20℃水化11~15h。
第一向 等电聚焦
1. 将纸电极置于聚焦盘的正负极上,加ddH2O 5~8µl润湿。
2. 取出水化好的胶条,提起一端将矿物油沥干,胶面朝下,将其置于刚好润湿的滤纸片上杂交以去除表面上的不溶物。
3. 将IPG胶条胶面朝下置于聚焦盘中,胶条的正极(标有+)对应于聚焦盘的正极,确保胶条与电极紧密接触。
4. 在每根胶条上覆盖2-3ml矿物油。
5. 对好正、负极,盖上盖子。设置等电聚焦程序。
6. 聚焦结束的胶条,立即进行平衡、第二向SDS-PAGE电泳。或将胶条置于样品水化盘中,-20℃冰箱保存,电泳前取出胶条,室温放置10分钟,使其溶解。
第二向SDS-PAGE电泳
1. 配制12%的丙烯酰胺凝胶。
2. 待凝胶凝固后,倒去分离胶表面的MilliQ水、乙醇或水饱和正丁醇,用MilliQ水冲洗。
3. 配制胶条平衡缓冲液I
4. 在桌上先放置干的厚滤纸,聚焦好的胶条胶面朝上放在干的厚滤纸上。将另一份厚滤纸用MilliQ水浸湿,挤去多余水分,然后直接置于胶条上,轻轻吸干胶条上的矿物油及多余样品,这样可以减少凝胶染色时出现的纵条纹。
5. 将胶条转移至样品水化盘中,加入6ml(17cm IPG)平衡缓冲液I,在水平摇床上缓慢摇晃15分钟。
6. 配制胶条平衡缓冲液II。
7. 第一次平衡结束后,取出胶条将之竖在滤纸上沥去多余的液体,放入平衡缓冲液II中,继续在水平摇床上缓慢摇晃15分钟。
8. 用滤纸吸去SDS-PAGE胶上方玻璃板间多余的液体,将二向凝胶放在桌面上,凝胶的顶部面对自己。
9. 将琼脂糖封胶液加热溶解。
10. 在100ml量筒中加入TGS电泳缓冲液。
11. 第二次平衡结束后,取出胶条,用滤纸吸去多余的平衡液(将胶条竖在滤纸上,以免损失蛋白或损坏凝胶表面)。
12. 用镊子夹住胶条的一端使胶面完全浸末在1×电泳缓冲液中漂洗数次。
13. 将胶条背面朝向玻璃板,轻轻放在长玻板上,加入低熔点琼脂糖封胶液。
14. 用适当厚度的胶片,轻轻地将胶条向下推,使之与聚丙烯酰胺凝胶胶面完全接触.注意:不要在胶条下方产生气泡,应推动凝胶背面的支撑膜,不要碰到胶面。
15. 放置5分钟,使低熔点琼脂糖封胶液凝固。
16. 打开二向电泳制冷仪,调温度为15℃。
17. 将凝胶转移至电泳槽中,加入电泳缓冲液,接通电源,起始时用的低电流(5mA~10mA/gel/17cm),待样品在完全走出IPG胶条,浓缩成一条线后,再加大电流(20-30mA/gel/17cm)待溴酚蓝指示剂达到底部边缘时即可停止电泳。
18. 电泳结束后,轻轻撬开两层玻璃,取出凝胶,并切角以作记号(戴手套,防止污染胶面)。
19. 进行染色。
SDS-PAGE胶染色
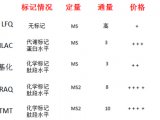
一、各种蛋白染色方法的灵敏度比较
染色方法 灵敏度 与质谱的兼容性 |
Silver 1-10ng - Silver (MS compatible) 10ng + Comassie G-250 or R-250 >30ng + Antibody (Western blot) 1ng - |
二、考马斯亮蓝染色
CBB染色液
0.5%考马斯亮蓝G250或R250
40%甲醇
10%乙酸
将CBB溶于甲醇中并不停的搅拌15min。加入乙酸与ddH2O
脱色液: 30%甲醇
10%乙酸
染色方法:1.在摇床上染色30min.
2.脱色至蛋白点或条带清晰可见
3. ddH2O洗3-5次
三、胶体考马氏亮蓝染色Colloidal Coomassie Staining(Cambridge centre for porteomics)
sensitivity = ~100ng
1. 固定:甲醇/醋酸/H2O(45:1:54)至少20min.
2. 染色12-18hr。
染色液: 17% (w/v) 硫酸铵
34%甲醇
0.5%醋酸
0.1% (w/v) Coomassie G250
3. 脱色:用H2O脱色至蛋白点和背景清晰.
双向电泳蛋白点的切取和保存
1. 用PDQuest软件或肉眼比对,找出感兴趣的蛋白点,并做好标记和记录。
2. 用MilliQ水冲洗胶2次。
3. 用色谱纯甲醇和MilliQ水冲洗Ep管
4. 将枪头(200µl)下端剪去,使其内径略小于蛋白斑点的直径,用色谱纯甲醇和MilliQ水冲洗枪头。
5. 对准斑点中央小心将蛋白切割下来,放入Ep管,MilliQ水漂洗2次,如胶块太大,将其切成1 x 1 mm的胶片。
6. 将切好的点做好标记和记录,置-80oC保存或冻干后-20oC存放。
注意事项:
1. 尽量避免皮肤和头发的角蛋白的污染,在操作过程中应戴一次性的PE手套(不用乳胶手套)和帽子。
2. 不要将胶长期存放于乙酸溶液中。
3. Ep管及染胶的容器必须用甲醇和水充分清洗,尤其应与进行Western blotting的容器分开,以避免casein或BSA的污染。
数据库搜索(Databases search)
一般来说,利用质谱数据鉴定蛋白质主要有两种方法:肽质量指纹图谱(PMF)和肽序列标签分析(sequence tag analysis)。这两种方法较传统的N端测序或内部Edman测序法敏感数百倍,其检测低限为飞摩尔(fmol)水平(如银染的2D胶点或1D胶带)。然而,足够多的样品蛋白量可以增加识别的成功率,这是因为增大样品蛋白量可以克服一些污染物的干扰(如角蛋白,在样品中的存在非常常见)。数据库的检索是利用计算机程序算法将实验测得的肽质量数据与蛋白质序列数据库中的蛋白质的肽质量计算值进行相关性比较分析,从而得出可能蛋白质的概率并将相关性最好的结果排序,这是凝胶分离蛋白质鉴定的最常用手段。
这种检索途径有一个明显的限制,就是被鉴定的蛋白质必须在序列数据库(蛋白质数据库和核酸序列翻译数据库)中存在。PMF检索鉴定蛋白质是依据实验中获得的蛋白质的多个肽段质量数据与同一蛋白质肽段质量理论计算值的之间的相关性比较。因此,这一技术不适用于检索EST翻译序列,也不适用于鉴定蛋白质的混合物。而肽序列标签分析(sequence tag analysis)可适用于检索EST数据库。
对PMF 数据,主要搜索Swiss Prot (速度快但不全面)和NCBI (速度虽慢但包含更多的信息)数据库。如果你认为你的蛋白质可能为新的蛋白质,选择NCBI数据库,标准搜索选择Swiss Prot 数据为好。如果需要,也可搜索EST数据库(用MS/MS数据按搜索更有效)。
检索条件的设置与结果判断:
1. 肽片段质量选择在800-4000Da范围。
2. 氨基酸残基的修饰(Modifications):半胱氨酸为脲甲基半胱氨酸(Cardamidomethyl-Cys),蛋氨酸选择可变修饰—氧化。
3. 最大允许的肽质量误差(Mass Tolerances),与仪器性能和数据质量有关,一般设为±0.1Da,最大为±0.5Da,质量误差愈小,搜索的特异性愈高。
4. 不完全酶解位点数目(Missed cleavages):每个肽允许有2个不完全裂解位点,一般选1(如果蛋白充分变性且酶解完全)。
5. 参考蛋白质表观分子量和等电点,表观PI误差范围为±0.5pH,表观Mr误差为范围为±20%。一般情况下不选此项,在判读结果时参考。
6. 物种选择:限定物种。
7. 离子选择:[M+H]+,单同位素
8. 最少匹配肽片段规定为4。
主要的公共数据库及网址






















 首页
首页


