近日,中国科学院大连化学物理研究所催化基础国家重点实验室理论催化创新特区研究组研究员肖建平团队发表了题为Toward computational design of chemical reactions with reaction phase diagram的综述文章,阐述了基于“反应网络全局搜索”的理论催化研究策略和基于“反应相图”的催化设计新方法。
对于具有“高维”复杂性的多相催化反应,可以通过建立一系列的线性关联,如吸附能之间的scaling关系,能垒和反应能之间的BEP关系等对其进行“降维”,并通过进一步的速率计算得到一系列催化剂表面催化活性的变化趋势,从而实现催化剂设计。传统理论催化设计方案主要为:吸附能之间线性关联的建立→能垒与反应能线性关联的建立→速率计算→催化剂设计。然而,由于反应网络的复杂性,传统催化活性趋势往往建立在假定催化路径的基础上。同时,过多的基元过程也会导致速率求解的困难。因此,在“理论先行”的催化剂设计中,这些因素都会提高设计成功的风险,影响其准确性。
在“反应相图”的理论催化研究的新范式中,该团队建立了特定催化反应的“全反应网络”,通过“反应网络全局搜索”的研究策略,可以得到不同反应相中可能出现的不同最优反应路径,由此建立第一级的热力学反应相图;进一步改进能垒和速率计算,得到更为准确的催化活性趋势,从而实现催化剂设计。“反应相图”的理论催化研究新范式主要为:反映网络的全局建立(线性关联的建立)→基于“反应网络全局搜索”的“反应相图”的建立→动力学“反应相图”的建立→催化剂设计。
该团队基于“反应网络全局搜索”和“反应相图”的研究策略,发现了二氧化碳电催化还原制甲酸的双顶点活性趋势(Nature Communications,2020;Advanced Materials,2021),深入讨论了氧还原过程中的电压对活性趋势的影响(The Journal of Physical Chemistry C,2020),以及合成气转化中的选择性问题(Chem,2020),并以“理论先行”成功设计了铜基电极材料进行一氧化氮电还原合成氨的催化过程(Angewandte Chemie International Edition,2020)。
该综述发表在WIREs Computational Molecular Science上。研究工作得到国家自然科学基金、中科院战略性先导科技专项(B类)“功能纳米系统的精准构筑原理与测量”和辽宁省“兴辽英才计划”等的资助。
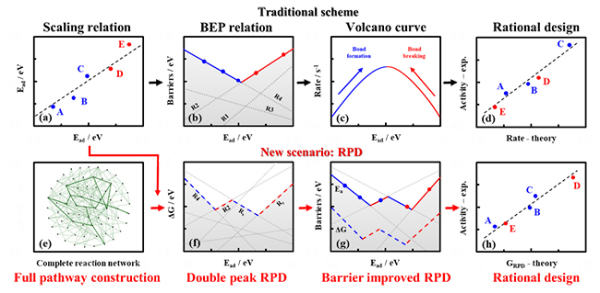
大连化物所发表基于“反应相图”的催化反应设计综述文章





 首页
首页

