【摘要】鸟枪法串联质谱蛋白质鉴定策略由于其高可靠和高效率而被广泛应用于蛋白质组学研究中,这种方法直接对蛋白质混合物进行酶切,以肽段为鉴定单元,继而推导真实的样品蛋白质。由于利用质谱图推导肽段存在一定的假阳性率,而且直接对蛋白质混合物的酶切也导致了肽段和蛋白质之间关联信息的丢失,所鉴定的蛋白质难免存在部分不可靠结果。 因此,蛋白质鉴定的质量控制在蛋白质组学研究中极为重要。蛋白质鉴定的质量控制包含两大类主要方法,其一为利用肽段进行蛋白质组装,当前最常用也被证明最有效的方法是使用简约原则,即用最少的蛋白质解释所有鉴定肽段,现有的方法可以分为布尔型和概率型,其二为鉴定蛋白质的可靠性评估,包括单个蛋白质鉴定置信度和蛋白质鉴定整体水平的假阳性率计算。综合各种可辅助蛋白质鉴定的先验信息,构建普适的概率统计模型,是目前蛋白质鉴定质量控制方法的发展趋势。
蛋白质组学研究最基本任务就是定性和定量地鉴定一个组织或细胞中的全部蛋白质[1, 2]。 质谱技术的快速发展使得在蛋白质组学水平上进行高通量的蛋白质鉴定成为可能。目前,生物质谱已成为大规模蛋白质组鉴定的核心技术,数据库搜索法是常用的图谱解析方法。尽管蛋白质鉴定的新算法不断涌现,由于质谱数据的复杂性和数据库搜索算法的局限性,肽段和蛋白质鉴定的质量控制问题十分突出,尤其体现在鸟枪法蛋白质组鉴定策略中[3]。随着仪器精度的提高和肽段鉴定质量控制算法的发展,肽段鉴定的精度越来越高,而蛋白质组鉴定的目的是鉴定出在细胞或组织中表达的蛋白质,因此,蛋白质水平的数据质量控制已成为蛋白质组鉴定中不可忽视的问题。
本文针对鸟枪法蛋白质鉴定的质量控制问题,将其分为蛋白质组装和蛋白质鉴定的可靠性评估两个方面介绍当前研究进展: 蛋白质组装是指利用鉴定肽段推导可能的样品蛋白,当前普遍使用简约原则进行推导,各种方法可以概括为布尔型和概率型组装法两个大类,蛋白质鉴定的可靠性评估包括单个蛋白质鉴定置信度和蛋白质整体水平假阳性率计算。蛋白质鉴定的可靠性评估对以质谱鉴定为核心的蛋白质组学研究具有重要的意义。
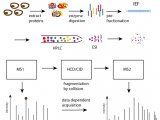
利用生物质谱鉴定蛋白质的过程分为实验和计算两步骤(图 1)。蛋白质样品经实验步骤获得质量图谱,经计算步骤进行图谱解析。
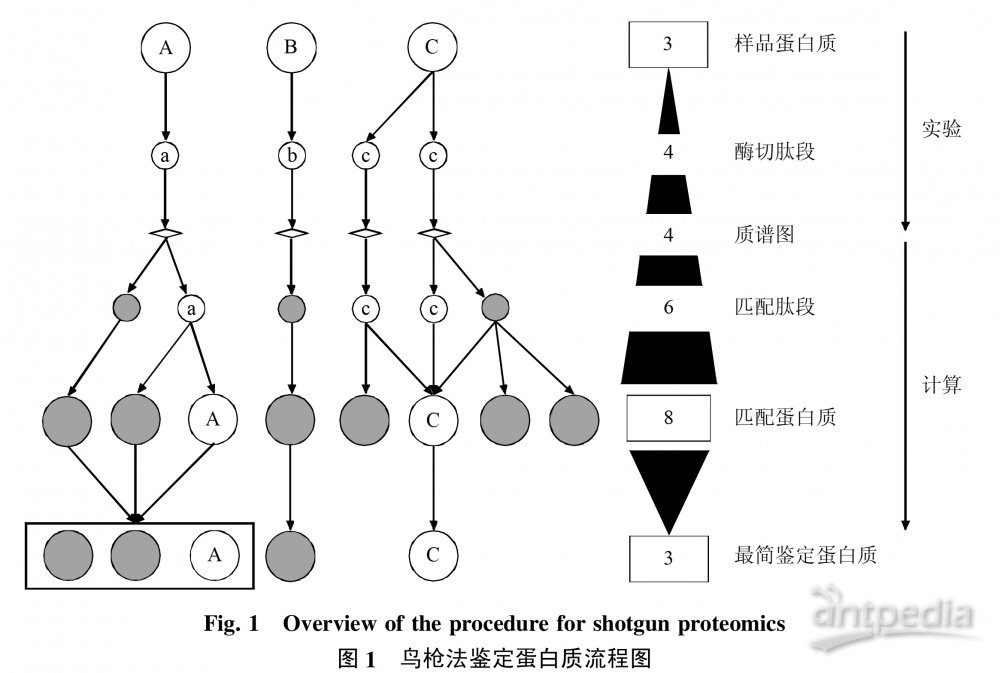
实验步骤可以归纳为两条技术线路:一条是自上而下(top-down)的技术路线,蛋白质需预先分离纯化(如二维凝胶电泳技术,2-DE),纯化后的蛋白质被酶切为肽段混合物,离子化后经一级质谱产生肽质量指纹图谱(peptide mass fingerprint, PMF),或经串联质谱产生肽碎片指纹图谱 (peptide fragmentation fingerprint, PFF)进行鉴定。另一条是自下而上(bottom-up)的技术路线,也称鸟枪法。鸟枪法的基本过程是,蛋白质混合物经过简单或不经过分离就被酶解为肽段混合物[4],肽段混合物经色谱分离和离子化后,经串联质谱产生 PFF 用于肽段鉴定,最后再从鉴定的肽段推导可能的蛋白质。
该方法可在短时间内获得大量鉴定结果,因此在蛋白质组研究中被广泛采用。
在计算步骤,现有的解析图谱方法包括序列库搜索、图谱库搜索、从头测序(de novo sequencing)以及从头测序结合容错性搜索的方法[5]。其中,蛋白质序列库搜索法是常用的解析图谱方法。一些经典的搜库软 件已得到广泛应用,其中包括MASCOT[6]、SEQUEST[7]、X!Tandem[8]等。 以鸟枪法技术路线为例,序列库搜索法的基本过程是:
a、将数据库中候选蛋白质序列理论酶切为肽段,模拟产生理论酶切肽段的碎裂图谱;
b、将理论图谱与实验图谱进行匹配,并据图谱相似性打分,经特定的肽段质量控制方法获得高可信的肽段鉴定结果;
c、根据肽段与蛋白质氨基酸序列的对应关系推导出蛋白质。
鸟枪法串联质谱鉴定的结果以肽段为鉴定单元,因而肽段鉴定的质量控制问题受到极大关注,当前常用的方法包括阈值过滤法、概率统计模型法等。概率统计模型法可计算出单个肽段鉴定的置信度,常 用 的 工 具包括PeptideProphet [9] 和PEP_PROBE[10]等。 近年来,随机库方法也被应用到大规模蛋白质组鉴定中,用于估算肽段鉴定结果的整体假阳性率或计算单个肽段鉴定置信度[11~13](本文所指的蛋白质鉴定的假阳性率都指鉴定结果中错误鉴定蛋白质所占的比例,肽段鉴定的假阳性率定义类同)。随着仪器精度的提高和肽段鉴定质量控制方法的逐渐成熟,如何从肽段推导出高可信的蛋白质,怎样对蛋白质进行可靠性评估等蛋白质鉴定质量控制问题逐渐受到关注。
鸟枪法蛋白质鉴定的质量控制主要体现在两方面:一是在单肽段匹配多个蛋白质时,判断哪个或哪几个更可能是样品中实际存在的蛋白质。
Nesvizhskii 等[14]指出,数据库特别是真核生物的数据库中存在大量相似序列的蛋白质,这其中包括来源于相同基因家族或可变剪接产生的同源蛋白质,以及测序错误、不同基因编号和功能注释造成的冗余序列等,因此,多个蛋白质共享单个或多个肽段的情况普遍发生,导致有时难以判断确切的来源蛋白质。二是蛋白质鉴定的可靠性评估。除了受共享肽段的影响,由于仪器(精度、噪声等)、图谱质量、搜库算法、数据库等因素造成肽段鉴定的假阳性,也会导致蛋白质鉴定的假阳性(图1)。建立合理的概率统计模型对所鉴定的蛋白质进行可靠性评估,如给出单个蛋白质鉴定的置信度以及大批量蛋白质鉴定结果的整体假阳性率,是蛋白质组鉴定数据质量控制的重要内容之一。近年来,计算蛋白质组领域陆续出现了有关蛋白质鉴定质量控制方面的工作,并陆续开发出一些工具(表1)。
2.1 蛋白质组装
为了解决由于共享肽段导致蛋白质的取舍问题,国际上逐渐形成了一种在简约原则下进行蛋白质组装的共识,且这种原则正被广泛应用于蛋白质推导中[14~22, 25, 27, 28]。简约原则也被称为奥卡姆剃刀原则(Occam’s Razor),即“用最少的蛋白质解释所有鉴定肽段”。2003 年,Nesvizhskii 等[25]首次将其应用到蛋白质置信度计算工具 ProteinProphet 中,不久 Yang 等[15]也使用了基于简约法的分类规则处理MASCOT 鉴定结果,随后简约原则在蛋白质推导中得到广泛应用,几乎所有蛋白质组装算法在处理共享肽段时都不同程度地使用了简约原则。2007年 Zhang 等[22]利用标准蛋白质数据,首次证明了简约原则下组装蛋白可以去掉样品中大量不存在的蛋白质,从而提高蛋白质鉴定的精确度。
目前,多种基于简约原则的算法被发展出来用于蛋白质组装(表 1)。有的方法可分为布尔型和概率型两类,前者将过滤后的肽段等同看待,根据简约原则推导出最简蛋白质列表,所鉴定蛋白质缺乏置信度打分。后者基于概率模型,可根据肽段置信度(简称肽段概率)给出每个蛋白质鉴定的置信度(简称蛋白概率),打分的结果会将不属于最简蛋白质的蛋白概率赋为 0[14, 25, 27]。无论哪类方法,由肽段组装蛋白质都可分为以下几步进行[5, 14]:
a.对经数据库搜索引擎(如 SEQUEST 或 MASCOT 等)鉴定的肽段进行过滤,按照一定规则获得高可信肽段鉴定结果;
b.从蛋白质序列库中找出肽段所匹配蛋白质的编号;
c.将匹配的蛋白质分组,不同组内的蛋白质之间无共享肽段;
d.每组内的蛋白质通过布尔型或概率型组装法给出最简蛋白质。
2.1.1 布尔型组装.
布尔型组装法是一类简单的基于简约原则的方法,其特点是经过过滤后的肽段被等同看待,推导的蛋白质无置信度打分。此类算法多采用启发式或贪婪算法。
DTASelect[19]是一个广泛使用的蛋白质组鉴定质量控制工具,在蛋白质组装时部分使用了简约原则,即将匹配到相同肽段集合的蛋白质合并为一个条目。数据库搜索软件 MASCOT 在给出蛋白质鉴定列表时也部分使用了简约原则,除了将具有相同肽段集合的蛋白质被合并为单个条目外,所属肽段之间是包含与被包含关系的蛋白质也合并作为一个鉴定结果(http://www.matrixscience.com/help/interpretation_help.html)。Yang 等将蛋白质按照匹配肽段之间的重叠关系划分为六类,在 DBParser[15]中对鉴定蛋白质进行简约分析,同样方法也被Slotta 等[16]运用到 MassSieve中对蛋白质进行去冗余。在 EPIR[17]中,具有相同肽段集合的蛋白质被当成一个单次鉴定条目,每次挑选包含肽段数最多的蛋白质,直至所有肽段被覆盖到。Isoform Resolver[18]中除采用贪婪算法获得最简蛋白质外,还考虑了氨基酸等质量或近质量的替换问题(如 I/L替换,K/Q 替换等),若存在等 / 近质量替换的肽段对应多个蛋白质,则挑选肽段覆盖率最高的蛋白质作为最终鉴定结果。Stephan 等在整合人类脑蛋白质组计划(human brain proteome project, HBPP)数据时,使用 ProteinExtractor[20, 21](整合在 ProteinScapeTM),挑选包含肽段数最多且可覆盖所有谱图的蛋白质。
Zhang 等[29]在处理小鼠血浆蛋白质数据时,优先选取了匹配图谱数和非共享肽段数最多的蛋白质。此外,某些计算蛋白概率的工具在处理共享肽段时,首先通过布尔型组装获得最简蛋白质,其次再计算蛋白概率[28]。
2007 年 Zhang 等[22]将蛋白质简约组装等效成二分图的最小集合覆盖问题[30],使用贪婪算法推导简约蛋白质集合,不仅首次证实了简约组装法可以提高蛋白质鉴定精度,而且评估了不同物种与数据库大小对蛋白质鉴定假阳性的影响,指出对搜索冗余度高和同源蛋白多的数据库获得的蛋白质进行简约组装更有利于提高蛋白质鉴定精度。
2.1.1 概率型组装.
除了布尔判决可获得最简蛋白质外,某些蛋白概率计算工具,如 ProteinProphet [25] 和PANORAMICS[27],也可用于简约组装。ProteinProphet可利用 PeptideProphet[9]或类似工具计算的肽段概率,在考虑了肽段所属蛋白质的其他肽段对该肽段概率的修正后,计算某蛋白质匹配的肽段中至少有一条被鉴定正确的概率,并通过期望最大化算法得到稳定的蛋白概率。其中若某肽段被多次重复鉴定,则取肽段概率最大值。迭代时,共享肽段的概率值在包含该肽段的所有蛋白质之间不断分配,直至趋于稳定,最终可使不属于最简蛋白质的蛋白概率分配为0 [14]。PANORAMICS 中 使 用 了 类 似ProteinProphet 的迭代过程,不同之处在于肽段鉴定概率由一个判别函数得到,且对氨基酸等 / 近质量替换的肽段合并为单个处理。
在将蛋白质组装归结为求解二分图的最小集合覆盖问题后,简约组装蛋白质在算法上实际上是一个 NP 难解问题[22, 30]。因此,尽管各种算法都采用了简约原则,但是对相同的数据集,算法实现上的差异性会产生不同的组装结果.此外,简约原则下推导蛋白质的整体假阳性率如何以及受哪些因素的影响,需要进行系统的分析和评估.这方面的工作除了 Zhang 等[22]曾比较了不同物种和数据库大小的影响,Padliya 等[31]也以植物病原体微生物为研究对象,研究了数据库中同源蛋白质对蛋白质鉴定精度的影响。
除在简约原则下进行蛋白质组装外,使用新的技术手段和利用生物学先验知识可提供更多信息,辅助蛋白质推导.例如鸟枪法结合定量蛋白质组学技术,使蛋白质推导可以借助肽段的定量信息。鸟枪法结合 2-DE 技术,可利用蛋白质的理化性质(如蛋白质分子质量)辅助蛋白质鉴定[20, 21]。利用肽段的可检测性[32, 33]和生物学先验知识(如 GO[34]或 HPRD[35]数据库的生物学注释)也可辅助蛋白质推导。
2.2 蛋白质鉴定可靠性评估
蛋白质鉴定的可靠性评估是蛋白质鉴定的最后一步,缺乏可靠性评估的结果不科学,是难以令人信服的.可靠性评估包括单个蛋白质鉴定置信度和蛋白质整体水平假阳性率计算两方面。
2.2.1单个蛋白质置信度计算
蛋白质鉴定的可靠性评估最好能给出每个蛋白质的鉴定置信度,即蛋白概率。蛋白概率包括蛋白质正确鉴定的概率以及由此衍生的指标,例如p 值、e 值等,旨在衡量某个蛋白质是否在样品中表达。现有的算法可根据先验肽段概率已知与否分为两类,已有工具见表 1。
a.肽段概率已知.
此类算法需已知肽段概率。现有方法包括ProteinProphet[25]的经验概率模型(蛋白概率为蛋白质所匹配肽段中至少有一条被正确鉴定的概率),PRO_PROBE[26]的二项分布模型以及 EBP[28]的贝叶斯概率模型。
ProteinProphet 的经验概率模型需已知肽段概率(由 PeptideProphet 或类似软件计算得到),通过期望最大化算法给出每个蛋白质正确鉴定的概率。然而该模型未能考虑数据集和数据库大小的影响。
Sadygov 等[10]在 PRO_PROBE 中将每次蛋白质鉴定看成一次贝努利事件(Bernoulli event),考虑了数据集和数据库大小等因素,采用似然比检验法得到蛋白质打分. 在 Sadygov 的模型中,H1 假设为肽段匹配到某蛋白质的次数符合二项分布,其中肽段概率从超几何分布模型计算得到,H0 假设为鉴定某蛋白质为一次随机匹配,匹配次数也符合二项分布,蛋白概率从数据库中蛋白质的相对长度(蛋白质的氨基酸与数据库中所有氨基酸个数之比)计算得到.在得到两种假设条件下的蛋白概率后,以这两种蛋白概率的似然比作为统计检验量来区分正确和错误鉴定,以似然比的自然对数作为蛋白质鉴定分数,统计检验的显著性水平可从模型的分布得到。
在大规模蛋白质组鉴定中,往往会采用多种数据分析路线(如使用多种数据库搜索引擎)或进行多次重复实验以提高蛋白质鉴定的精度。Price 等[28] 基于贝叶斯概率模型开发了 EBP,用于整合多种搜库引擎和多次重复实验的鉴定结果。在 EBP 模型中,肽段概率首先由 PeptideProphet 计算得到,并通过一个函数整合多个搜库引擎鉴定的肽段概率(返回肽段鉴定结果一致的最大概率,对不一致的肽段鉴定进行罚分)。得到整合后的肽段概率后,假设正确和错误的肽段鉴定在“肽段空间”随机发生并受多种因素控制(蛋白质长度、蛋白质丰度、数据集和数据库大小等),这些因素被考虑进一个似然函数用于估计蛋白概率以及更新模型参数,并通过期望最大化算法使似然函数最大化,进而得到蛋白质正确鉴定的概率。Price[28]和 Lucitt[36]等都利用 EBP 鉴定到上千个高可信的斑马鱼蛋白质,随机库方法估计其假阳性率都在 1%以下。
b.肽段概率未知.
此类算法一般先根据图谱匹配打分、假设分布或判别函数等方法估计出肽段概率或肽段随机匹配p 值,其次再根据肽段随机匹配到某蛋白质次数的假设分布(如二项分布、多项分布和泊松分布)或类似 ProteinProphet 的经验概率模型计算蛋白概率。
适用于分析不同搜库引擎生成数据的模型包括Qscore 模型[23]、多项分布模型[26]、泊松分布模型[37]以及随机抽样模型[13]等。Moore 等在 Qscore 中采用近似二项分布模型计算蛋白概率,考虑的因素包括鉴定肽段数、单个蛋白质匹配肽段数、数据库大小和肽段概率(1 减去实际图谱与理论图谱归一化点积差值的倒数),最后的分值取蛋白质随机匹配概率的负对数值。Sadygov 等发展了另一种无需知道先验概率(肽段概率)的多项分布模型,用于计算SEQUEST 搜库结果的蛋白概率。该模型首先统计出肽段鉴定的 p 值随 Xcorr 值的分布,蛋白概率就是观察到一组特定的肽段打分的多项分布概率。由于所计算的蛋白概率基于 p 值,可等同为蛋白质随机匹配的概率。同 Qscore 一样,该方法同样可利用多种搜库引擎的肽鉴定打分。States 等[37]认为肽段随机匹配次数符合泊松分布,由此推出具有特定长度的蛋白质随机匹配概率,经 Bonferroni 校正的多重检验可得到蛋白质随机匹配的期望值.利用此方法他们对人类血浆蛋白质组计划(human plasma proteome project, HPPP)的数据重新分析,从 9,504个蛋白质中找出了 889 个置信度大于或等于 95%的蛋白质。Ramos-Fernandez 等[13]则另辟蹊径,在得到蛋白质鉴定打分后,进一步生成蛋白质随机匹配分值分布,用于估计蛋白质鉴定 p 值.他们首先根据单个蛋白质的肽段匹配数 h 将蛋白质进行分类,并对每类蛋白质分别进行 106 抽样,每次抽样计算出一个蛋白质打分 Sp:即随机抽取 h 个匹配到随机数据库的肽段 p 值,将这 h 个 p 值的负对数值相加作为蛋白打分 Sp.这样不同类的蛋白质会有一个对应的随机匹配分值分布,某个 Sp 对应的 p 值就是大于或等于该分值的蛋白质的相对频率。其中肽段 p 值计算利用了广义 Lambda 分布(generalized Lambda distribution)拟合匹配到随机库的肽段分值分布得到。
针对 MASCOT 鉴定结果,Feng 等[27]开发了PANORAMICS 用来计算蛋白概率.该算法首先从MASCOT 鉴定肽段的离子分数(ion score)出发,在考虑了数据库大小和母离子质量误差容限下候选肽段数目对肽段概率的影响后,给出一个计算肽段概率的线性函数,并使用标准蛋白质数据,通过线性回归进行参数估计。得到肽段概率后,计算蛋白概率的方法则与 ProteinProphet 类似,且考虑了不同电荷状态对肽段概率的影响.该方法计算时间复杂度低,在处理大规模数据集和搜索数据库的选择上具有很好的优势,缺点在于只能处理 MASCOT 的搜库结果。
综上所述,蛋白概率计算方法的发展大致有如下趋势:一是更广的适用性,包括不依赖已有的肽段打分,而在模型中利用先验知识计算肽段概率,例如Qscore中的肽段概率计算方法,RamosFernandez 等的广义 Lambda 分布拟合模型等,都可适用于多个搜库引擎的肽段打分系统.EBP 虽然需要 PeptideProphet 计算肽段概率,但当二者被整合到串联质谱数据分析软件 TPP (http://tools.proteomecenter.org/TPP.php) 中后,PeptideProphet很广的适用性弥补了这个缺陷。此外,更广的适用性体现在可整合源自多个搜库引擎和多次重复实验的鉴定结果。二是更多影响因素被考虑到蛋白概率计算模型中,以上模型考虑因素可以概括为 9 个参数:蛋白质长度、蛋白质丰度、数据集大小、数据库大小、与母离子具有相似 m/z 的候选肽段数目、单个蛋白质的匹配肽段数、肽段重复鉴定次数、肽段带电量和肽段概率。考虑更全面的影响因素,构建更合理的概率模型,会产生更理想的蛋白概率。
2.2.2 蛋白质鉴定整体假阳性率估计.
在蛋白质鉴定的可靠性评估中,除了计算单个蛋白质鉴定置信度外,降低鉴定结果整体假阳性率,对假阳性率进行准确估计也是非常重要的一个方面。卡多肽段[38]和采用多个搜库引擎[20, 21, 39]的方法常被用于降低结果的假阳性率,如 HPPP 的数据分析取双肽段或以上(大于或等于两个非冗余肽段)的蛋白质作为鉴定结果[38],HBPP[20, 21]中则使用了多个搜库引擎鉴定蛋白质。另外,Rohrbough 等[39]提出可用多个搜库引擎来验证单肽段鉴定蛋白质的置信度。在计算蛋白质鉴定整体假阳性率时,除ProteinProphet 中采用蛋白质错误鉴定概率的平均值进行估计外,也有利用泊松模型[38]和随机库方法[12, 40]进行估算的。HPPP 数据分析中使用了泊松模型[38]估计蛋白质鉴定整体假阳性率:假定肽段随机匹配次数满足泊松分布,从单肽段匹配蛋白质的高可信鉴定数和总鉴定数可得到单肽段水平上蛋白质鉴定的真阳性率,再利用此真阳性率计算泊松模型中的肽段随机匹配频率 姿,进而获得蛋白质在不同肽段数水平上的总体假阳性率。
利用随机库方法计算肽段鉴定结果假阳性率已有广泛应用[11, 12],而估计蛋白质整体水平假阳性率的应用则相对较少.随机库方法用于估计蛋白质鉴定的假阳性率时,一般假设匹配到随机库里的蛋白质个数即为正库里随机匹配的蛋白质个数。Weatherly 等[40]将蛋白质按照肽段匹配数划分,通过搜索反库(构造随机库方法之一,即将正库里氨基酸序列直接反转)计算每类蛋白质鉴定的假阳性率.Stephan 等[20, 21]在整合 HBPP 结果时,使用蛋白质鉴定假阳性率不能超过 5%作为约束条件,其中假阳性率估计使用了反库方法.此外,在某些蛋白概率算法的评估中,也使用了随机库方法估计的假阳性率作为经验标准[27, 28]。实际上,随机库方法估计的假阳性率是否能代表蛋白质鉴定的真实假阳性率,目前仍存在争议.Elias 等[12]指出,将随机库方法应用于蛋白质假阳性率的估计时,会导致正确鉴定蛋白质数的高估。
2.2.3 蛋白质鉴定可靠性评估方法比较.
对各种蛋白质鉴定可靠性评估方法的比较一般可通过构建模拟数据,或者随机库的方法计算鉴定结果的假阳性率或假阴性率,并以此作为标准衡量方法的优劣.另外构建模拟数据集还可评估其他因素的影响,如数据集和数据库大小等。
Xue 等[41]利用半随机抽样模型模拟搜库的肽段鉴定结果,比较了 4 种蛋白质可靠性评估方法:
ProteinPropht[25]算法核心公式,PROT_PROBE[26]的二项分布模型,HPPP 的泊松模型[38]以及取两肽段或以上蛋白质的方法。评估结果表明:PROT_PROBE 的二项分布模型能较好地区分鉴定结果中假阳性和真阳性蛋白质;ProteinProphet 算法核心公式计算的蛋白概率高于真实结果且区分度不好;HPPP 所采用的泊松模型,在一定程度上较准确地计算假阳性蛋白质鉴定数,但需要预先知道单肽段鉴定结果的可靠性,而且没有考虑蛋白质长度的影响;卡双肽段的方法则是比较有效却有些粗略的方法,蛋白质鉴定精度易受到数据集和数据库大小的影响. 另外,Price 等[28]用随机库方法估计的假阳性率比较了 EBP 与 ProteinProphet,指出EBP 的概率估计相对保守,ProteinProphet 则会引入更多假阳性结果。Feng 等[27]也使用了随机库方法比较了 PANORAMICS 与 ProteinProphet,发现与PANORAMICS 相比,ProteinProphet 在概率接近 1时具有相对较低的假阳性率,但是全部概率范围内却具有较低的精度,并指出原因在于 ProteinProphet引入的肽段概率修正使肽段匹配蛋白的独立假设不再成立,因而原经验概率公式不再适用。
综上所述,各种蛋白质鉴定的可靠性评估法各有优点,也各有缺陷。整合多种先验信息和影响因素,建立通用的概率统计模型,给出每个蛋白质的鉴定概率是较好的选择,但模型的验证仍面临缺乏大批量标准蛋白数据集的困难。
蛋白质鉴定是蛋白质组学研究的基础,只有鉴定到生物样品中真实表达的蛋白质,才能准确获得蛋白质相互作用、亚细胞定位、蛋白质修饰等信息。但是,受样品、实验设计和仪器等因素限制,质谱鉴定数据存在着严重的质量问题,这就对后续的质量控制算法提出了很高的要求。鸟枪法鉴定结果首先获得鉴定肽段,随后再获得鉴定蛋白质。有研究表明,即使能获得较高可靠性的鉴定肽段,其推导的蛋白质可靠性也可能会低得多.随着质谱仪器精度的提高和肽段鉴定数据质量控制方法的不断完善,蛋白质鉴定数据质量控制问题日趋显著。本文首次将蛋白质鉴定质量控制明确分为蛋白质组装和蛋白质鉴定的可靠性评估两部分,并对其进展情况进行了系统综述.随着人类蛋白质组计划的不断实施,数据挖掘的深度和广度也越来越大,制定统一的数据标准和数据分析流程,发展普适的概率统计模型辅助蛋白质鉴定的可靠性评估已成为趋势。
致谢 感谢北京蛋白质研究中心生物信息学研究室荔建琦博士和侯林博士生在算法上的讨论。
生物化学与生物物理进展,Progress in Biochemistry and Biophysics 2009, 36(6): 668~675 。李 宁 吴松锋 朱云平 ** 杨晓明
参 考 文 献
1 Aebersold R, Mann M. Mass spectrometry-based proteomics.Nature, 2003, 422 (6928): 198~207
2 He F. Human liver proteome project: plan, progress, and perspectives. Mol Cell Proteomics, 2005, 4 (12): 1841~1848
3 Steen H, Mann M. The ABC忆s (and XYZ忆s) of peptide sequencing. Nat Rev Mol Cell Biol, 2004, 5 (9): 699~711
4 Yates III J R. Mass spectral analysis in proteomics. Annu Rev Biophys Biomol Struct, 2004, 33: 297~316
5 Nesvizhskii A I, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods, 2007, 4 (10): 787~797
6 Perkins D N, Pappin D J, Creasy D M, et al. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis, 1999, 20 (18): 3551~3567
7 Eng J K, McCormack A L, Yates III J R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a proteins database. J Am Soc Mass Spectrom, 1994, 5 (11): 976~989
8 Craig R, Beavis R C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics, 2004, 20 (9): 1466~1467
9 Keller A, Nesvizhskii A I, Kolker E, et al. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem, 2002, 74 (20): 5383 ~5392
10 Sadygov R G, Yates III J R. A hypergeometric probability model for protein identification and validation using tandem mass spectral data and protein sequence databases. Anal Chem, 2003, 75 (15): 3792~3798
11 Elias J E, Haas W, Faherty B K, et al. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods, 2005, 2 (9): 667~675
12 Elias J E, Gygi S P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods, 2007, 4 (3): 207~214
13 Ramos-Fernandez A, Paradela A, Navajas R, et al. Generalized method for probability-based peptide and protein identification from tandem mass spectrometry data and sequence database searching. Mol Cell Proteomics, 2008, 7 (9): 1748~1754
14 Nesvizhskii A I, Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Prot, 2005, 4 (10):1419~1440
15 Yang X, Dondeti V, Dezube R, et al. DBParser: web-based software for shotgun proteomic data analyses. J Proteome Res, 2004, 3 (5):1002~1008
16 Slotta D J, McFarland M, Makusky A, et al. P18-T Mass Sieve: a new tool for mass spectrometry-based proteomics. J Biom Tech, 2007, 18 (1): 7
17 Kristensen D B, Brond J C, Nielsen P A, et al. Experimental peptide identification repository (EPIR): an integrated peptide-centric platform for validation and mining of tandem mass spectrometry data. Mol Cell Proteomics, 2004, 3 (10): 1023~1038
18 Resing K A, Meyer-Arendt K, Mendoza A M, et al. Improving reproducibility and sensitivity in identifying human proteins by shotgun proteomics. Anal Chem, 2004, 76 (13): 3556~3568
19 Tabb D L, McDonald W H, Yates III J R. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res, 2002, 1 (1): 21~26
20 Stephan C, Reidegeld K A, Hamacher M, et al. Automated reprocessing pipeline for searching heterogeneous mass spectrometric data of the HUPO brain proteome project pilot phase. Proteomics, 2006, 6 (18): 5015~5029
21 Hamacher M, Apweiler R, Arnold G, et al. HUPO brain proteome project: summary of the pilot phase and introduction of a comprehensive data reprocessing strategy. Proteomics, 2006, 6 (18): 4890~4898
22 Zhang B, Chambers M, Tabb D. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J Proteome Res, 2007, 6 (9): 3549~3557
23 Moore R E, Young M K, Lee T D. Qscore: an algorithm for evaluating SEQUEST database search results. J Am Soc Mass Spectrom, 2002, 13 (4): 378~386
24 Kislinger T, Rahman K, Radulovic D, et al. PRISM, a generic large scale proteomic investigation strategy for mammals. Mol Cell Proteomics, 2003, 2 (2): 96~106
25 Nesvizhskii A I, Keller A, Kolker E, et al. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem, 2003, 75 (17): 4646~4658
26 Sadygov R G, Liu H, Yates J R. Statistical models for protein validation using tandem mass spectral data and protein amino acid sequence databases. Anal Chem, 2004, 76 (6): 1664~1671
27 Feng J, Naiman D Q, Cooper B. Probability model for assessing proteins assembled from peptide sequences inferred from tandem mass spectrometry data. Anal Chem, 2007, 79 (10): 3901~3911
28 Price T S, Lucitt M B, Wu W, et al. EBP, a program for protein identification using multiple tandem mass spectrometry datasets. Mol Cell Proteomics, 2007, 6 (3): 527~536
29 Zhang Q, Menon R, Deutsch E, et al. A mouse plasma peptide atlas as a resource for disease proteomics. Genome Biology, 2008, 9 (6):R93
30 Cormen T H, Leiserson C E, Rivest R L, et al. Introduction to Algorithms. 2nd. Cambridge: MIT Press and McGraw-Hill, 2001.1033~1039
31 Padliya N D, Garrett W M, Campbell K B, et al. Tandem mass spectrometry for the detection of plant pathogenic fungi and the effects of database composition on protein inferences. Proteomics, 2007, 7 (21): 3932~3942
32 Tang H, Arnold R J, Alves P, et al. A computational approach toward label-free protein quantification using predicted peptide
detectability. Bioinformatics, 2006, 22 (14): e481~e488
33 Alves P, Arnold R J, Novotny M V, et al. Advancement in protein inference from shotgun proteomics using peptide detectability. Pac Symp Biocomput, 2007. 409~420
34 Ashburner M, Ball C A, Blake J A, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium Nat Genet, 2000, 25 (1): 25~29
35 Peri S, Navarro J D, Kristiansen T Z, et al. Human protein reference database as a discovery resource for proteomics. Nucl Acids Res, 2004, 32 (suppl_1): D497~D501
36 Lucitt M B, Price T S, Pizarro A, et al. Analysis of the zebrafish proteome during embryonic development. Mol Cell Proteomics, 2008, 7 (5): 981~994
37 States D J, Omenn G S, Blackwell T W, et al. Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nat Biotechnol, 2006, 24 (3): 333~338
38 Adamski M, Blackwell T, Menon R, et al. Data management and preliminary data analysis in the pilot phase of the HUPO plasma proteome project. Proteomics, 2005, 5 (13): 3246~3261
39 Rohrbough J G, Breci L, Merchant N, et al. Verification of single-peptide protein identifications by the application of complementary database search algorithms. J Biomol Tech, 2006, 17 (5): 327~332
40 Weatherly D B, Astwood III J A, Minning T A, et al. A Heuristic method for assigning a false-discovery rate for protein identifications from Mascot database search results. Mol Cell Proteomics, 2005, 4 (6): 762~772
41 Xue X, Wu S, Wang Z, et al. Protein probabilities in shotgun proteomics: evaluating different estimation methods using a semi-random sampling model. Proteomics, 2006, 6 (23): 6134 ~6145















































 首页
首页


