前 言
距离2020年还有12个工作日,您是否还在为2019年未完成的科研小目标而烦恼?如果您之前做了多个组学的分析,面对海量数据不知如何深入挖掘,亦或是手头有其他临床或生理指标想与多组学数据结合,而不知如何下手,那么本篇文章绝对是您逆袭的法宝。
今天小欧为大家重点介绍一款探索多组学数据的软件Multi-Omics Factor Analysis(以下简称MOFA),助力广大奋战在科研第一线的小伙伴在2019年最后一个月冲刺。
我们先来看看欧易生物MOFA的分析流程吧:
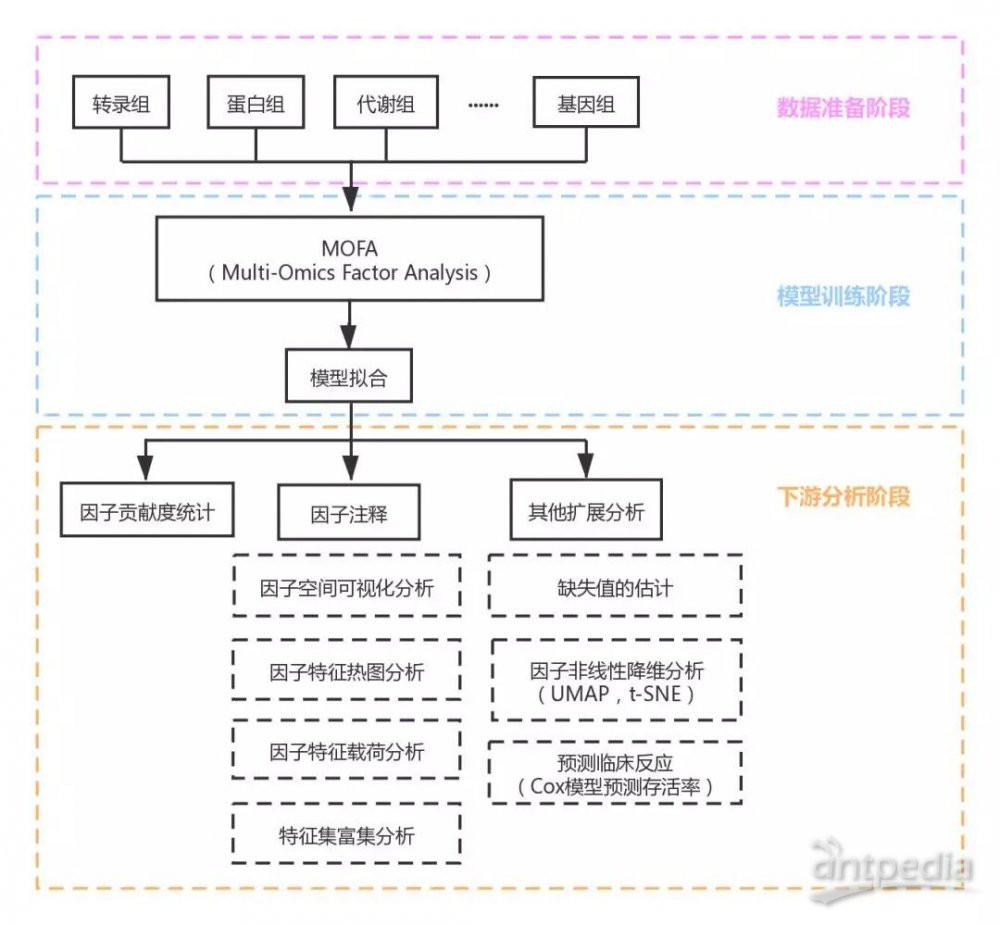
图 欧易生物MOFA分析流程图
如上图所展示,MOFA分析可以在常规组学分析基础上,进一步将数据进行整合,从因子分析的角度解析影响不同组学数据背后异质性因素,随后可对解析出的因子进行注释及其他扩展分析。MOFA软件相比于传统联合分析软件有比较明显的优势:
MOFA软件优势
样本量:既适用于大样本量数据,也适用于小样本量数据(每个组学具有一一对应关系的样本至少15个以上);
缺失值估计:对于有缺失值的样本,可以进行缺失值的估计;
适用的数据类型广泛:可对离散型数据、连续型数据和二进制数据进行分析;
应用范围广:适用于两个及两个以上组学,因子分析结果还可以继续进行非线性降维聚类(t-SNE,UMAP)和预测临床反应(Cox模型)等分析;
分析角度独特:从因子贡献度角度解析数据异质性,为多组学研究提供了新思路。
看到了如此多的优点,老师们是不是更加迫不及待了?接下来我们就从分析原理和分析内容展示两方面来具体介绍~
1
MOFA分析原理

图1 MOFA分析原理
MOFA是一种以无监督方法,集成多个模态组学数据的分析方法。分析流程主要可以分为两个步骤:第一步,对于给定n(n>=2)个数据矩阵,MOFA可以在完全重叠或部分重叠的样本集中识别多种组学数据类型,推断影响数据集模态主要变异来源的潜在因子。第二步,通过模型训练的输出结果可以用于一系列下游分析,包括因子空间可视化,因子载荷分析、因子聚类分析以及使用(基因集)富集分析对因子进行注释和缺失值的估计。
2
分析内容展示
在了解MOFA原理后,我们来结合实际模拟数据具体来进一步了解下MOFA软件的分析内容。
2.1数据展示
本次研究对象是170 例人类样本,对以上样本分别进行转录组、代谢组和微生物多样性测序,得到的标准化后的表达矩阵作为MOFA的输入文件。输入数据基本展示情况如下:
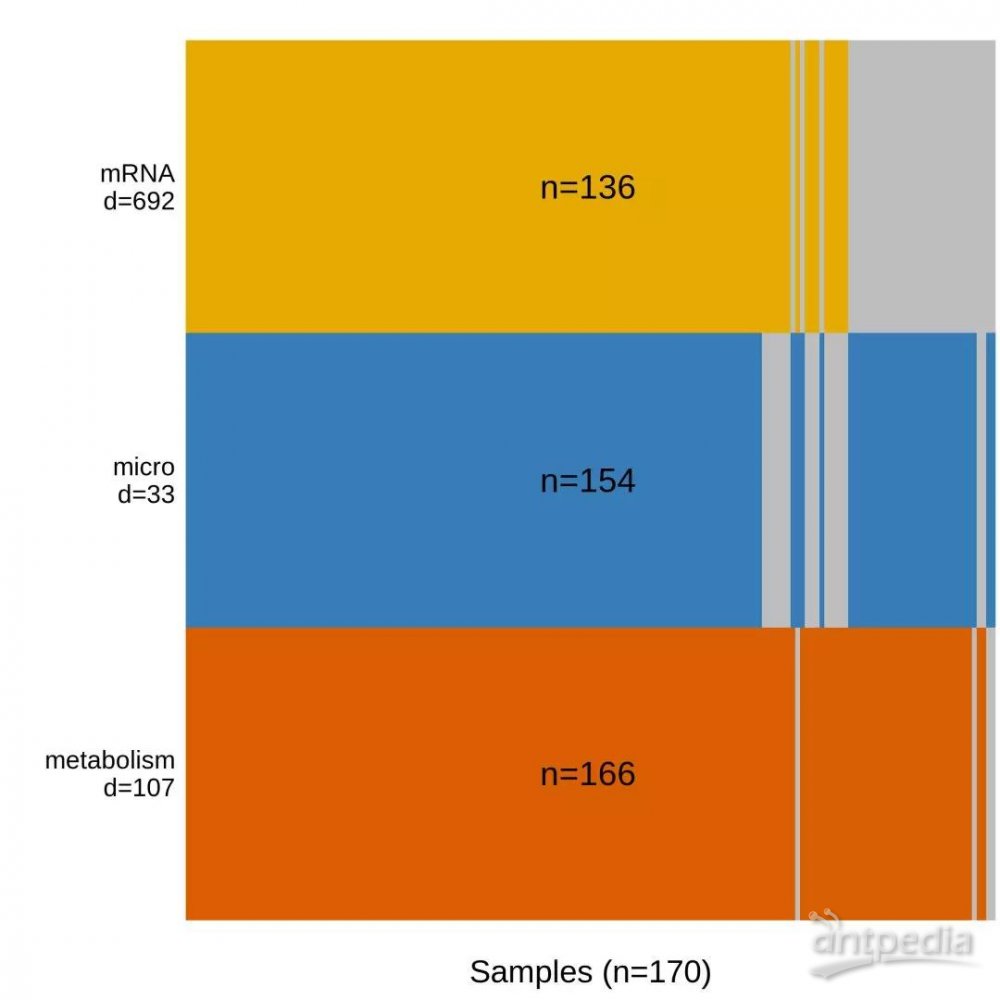
图2.1 数据基本情况展示
其中横轴(n)代表样本数据,纵轴代表输入的数据来源及样本特征(d),灰色代表缺失样本。
2.2 模型预测
上述数据输入MOFA软件进行模型训练,可将异质性因素分解为少数潜在因子,并统计各个因子的贡献度以及在各个数据来源中的累积情况,统计如下图所示:

图2.2 各数据中潜在因子贡献累积情况(上图)以及潜在因子贡献度(下图)展示
其中上图横轴代表不同的组学,纵轴代表各个因子在不同组学中的累积贡献度;下图横轴代表不同的组学,纵轴代表各个潜在的影响因子,颜色代表贡献度的高低,颜色越深代表贡献度越高。即从数据来源角度,累积贡献度从高到底排序为,微生物多样性( 60.8 %),转录组(50.7%)和代谢组(22.7 %),其中微生物多样性数据中因子1和因子4贡献度较高,转录组数据中因子3和因子5贡献度较高,代谢组数据中因子2和因子7贡献度较高。
2.3 因子分析
因子仍然比较抽象,那么解析出贡献度高的因子后,如何将重要因子和我们的基因、代谢物、微生物数据相关联呢?在了解各个数据源潜在影响因子贡献度后,接下来可以对每个来源中的贡献度靠前的因子及其特征进行重点分析。即找到影响因子贡献度最重要的那些基因、代谢物、微生物。
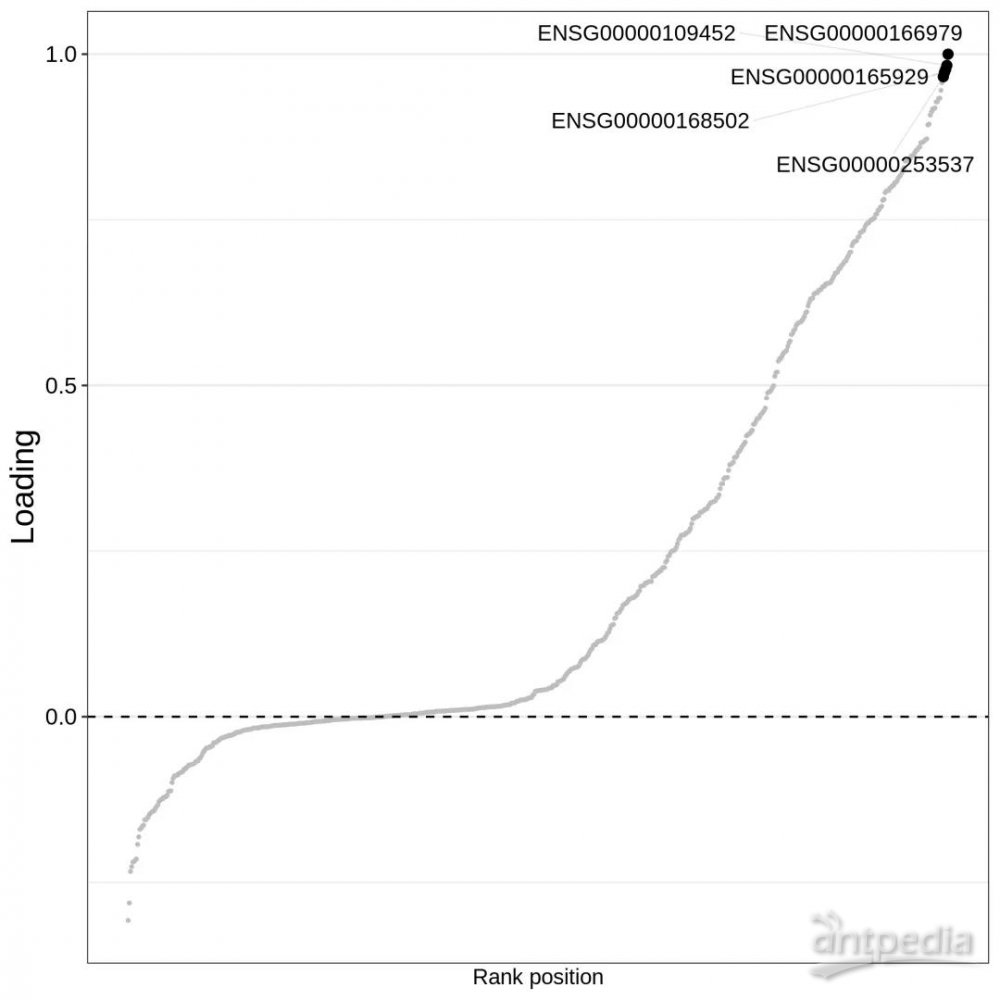
图2.3.1 转录组数据中因子3的绝对载荷图
其中纵轴代表绝对载荷,横轴为根据绝对载荷数值大小的排序。上图以转录组数据因子3水平的绝对载荷为例,并同时对载荷位于前五的基因进行名称标注。特别地,也可以对排名靠前的基因载荷进行重点展示,如下图:
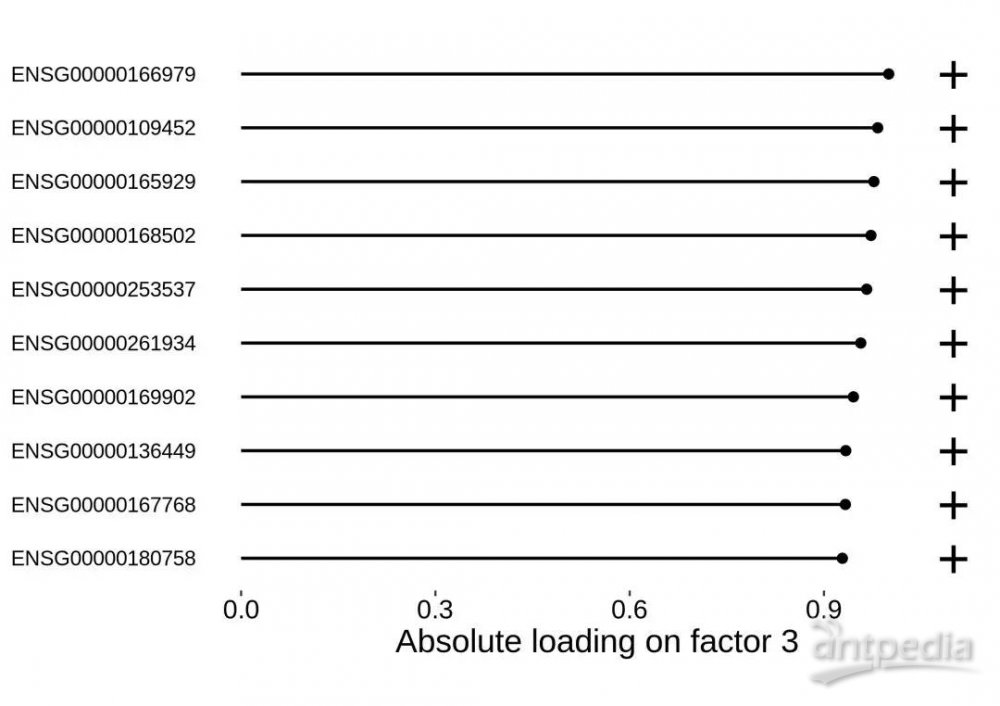
图2.3.2 转录组数据因子3水平上数据权重top10 特征载荷图
其中上图横轴代表载荷比例,纵轴代表转录组数据中的基因名称。以转录组数据因子3水平上占比最大权重的前10个基因载荷为例,载荷数据越大代表对该因子的贡献度越大。
2.4 富集分析
对于贡献度高或是感兴趣的因子(可以是任意组学角度),以转录组为例,可以继续探索其所在的通路来获知具体的功能,这里MOFA软件提供了GSEA富集分析(仅针对人和小鼠物种)。
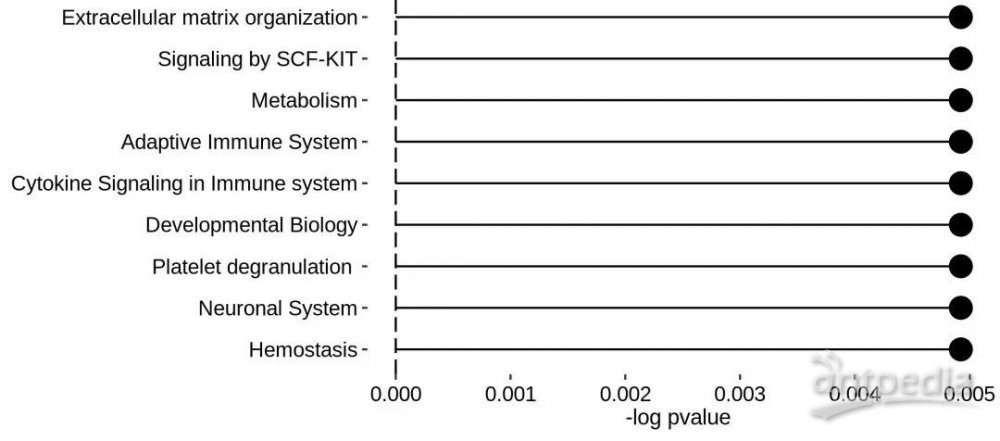
图2.4.1 MOFA 软件GSEA富集分析
其中纵轴为富集到的通路名称,横轴代表-log pvalue值,其值越高,代表pvalue值越小,该通路越显著。
当然,如果是这两个物种之外的其他物种,也可以用欧易的GSEA流程分析。

图2.4.2 欧易生物 GSEA富集分析
上图分成3个部分,第一部分为基因Enrichment Score的折线图,横轴为该基因下的每个基因,纵轴为对应的Running ES, 在折线图中有个峰值,该峰值就是这个基因集的Enrichment score,峰值之前的基因就是该基因集下的核心基因。第二部分为hit,用线条标记位于该基因集下的基因,第三部分为所有基因的rank值分布图,采用signal_to_noise 算法。从该图中可以看出,这个基因集在Case 组高表达。
另外,还可以通过热图来展示位于该基因集下的基因在所有样本中表达量的分布,示例如下:
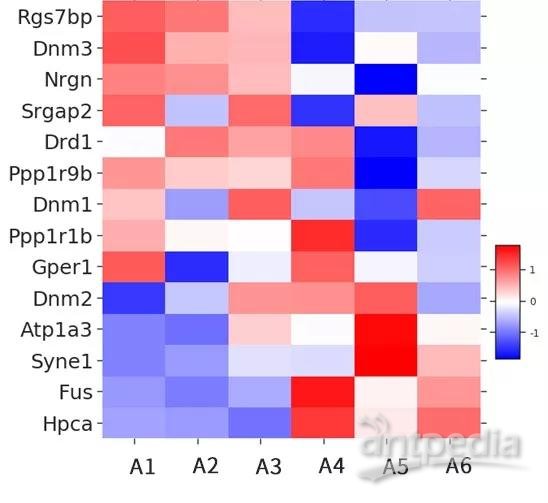
图2.4.3 指定基因集下的基因在所有样本中表达量热图
其中每一列代表一个样本。每一行代表一个基因,基因表达量从低到高,颜色从蓝色过渡到红色。
除了可以对转录组数据的因子特征(贡献度高的基因)进行分析,也可以对代谢组(贡献度高的代谢物)或者微生物(贡献度高的微生物)的因子特征进行分析。以微生物多样性为例,在确定了某个因子后,可以分析出对该因子贡献度最高的Top30物种,拿到这些物种列表后,基于物种丰度,可以进一步进行“因子高贡献度”物种的相关性热图分析或者物种相关性network分析。
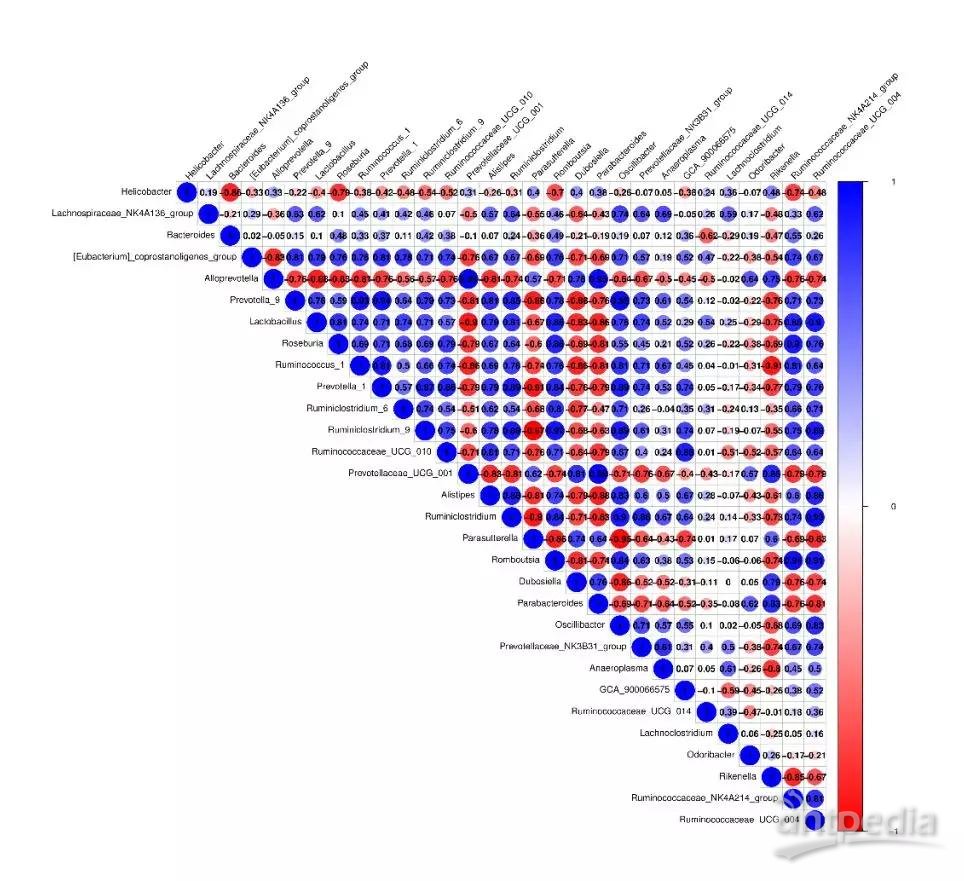
图2.4.4 “因子高贡献度”物种相关性热图分析
其中红色为负相关,蓝色为正相关,颜色越深,圈越大,则相关性越强。
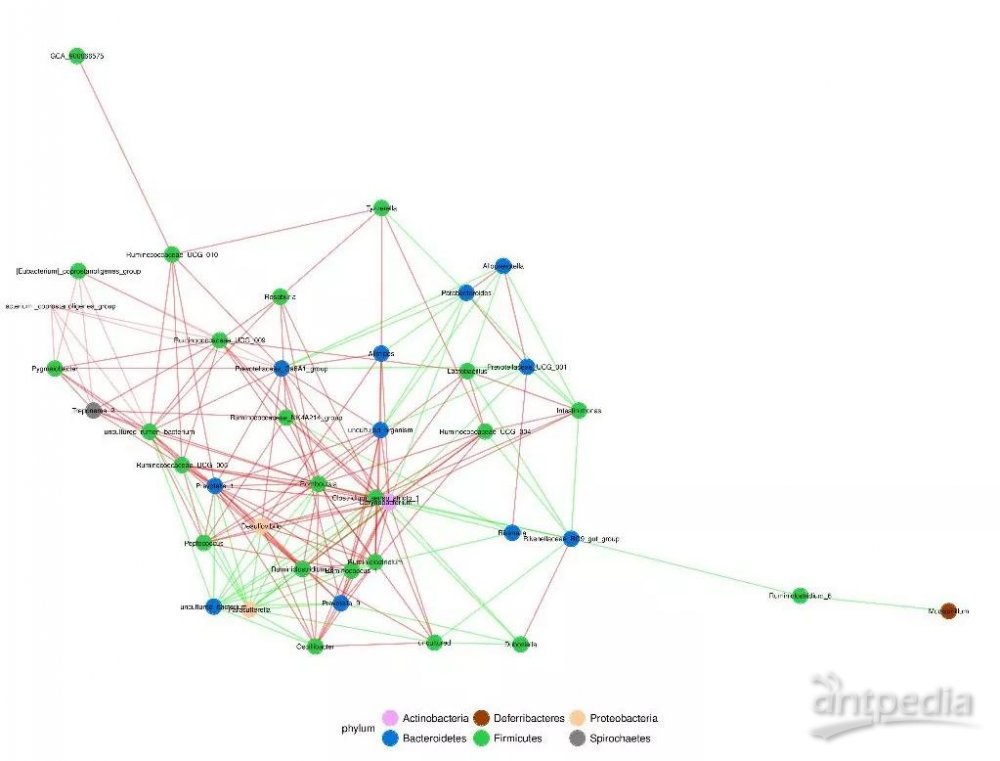
图2.4.5 物种相关性network图
其中节点的大小表示物种丰度的大小,不同颜色表示不同的物种;连线的颜色表示正负相关性,红色表示正相关,绿色表示负相关;线的粗细表示Pearson相关性系数的大小,线越粗,表示物种之间的相关性越高;线越多,表示该物种与其他物种之间的联系越密切。
2.5 样本聚类分析
同时,也可以基于感兴趣的因子对所有样本进行K-means聚类分析(本案例测试数据设置k=3)。

图2.5.1 转录组数据所有样本在因子1到因子5 样本K-means聚类展示
从整体来看,横轴和纵轴为因子1到因子5的样本聚类情况,每一个小的聚类图为两两因子作用下样本聚类情况,图例中1,2,3分别代表K-means聚类中的三个cluster,NA代表转录组中存在缺失的样本。上图展示了不同因子两两相互作用下,转录组样本的整体分类情况。
MOFA软件还可以通过添加外部指标数据(比如临床指标数据。数据可以是分类变量,也可以是数值型变量),来对样本进行分类。我们在2.2中提到微生物数据中因子1的贡献度最大(为40%),在因子1水平上,导入外部指标数据后,通过Beeswarm图进行可视化。
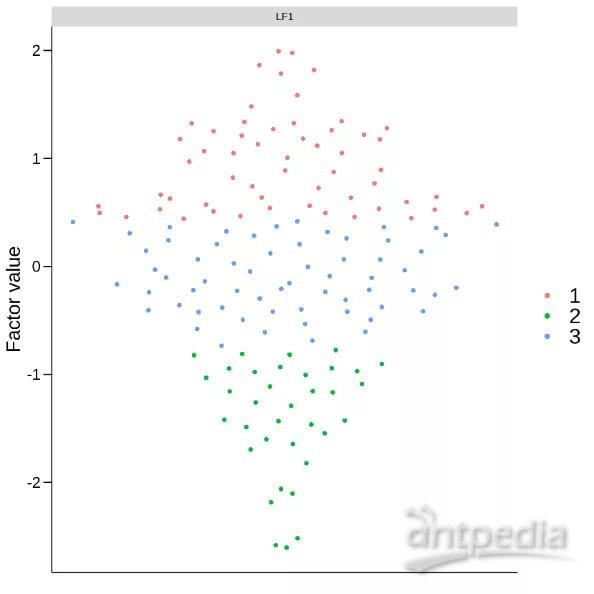
图2.5.2 微生物数据样本在因子1水平的Beeswarm图形展示
上图中可以看出根据纵坐标factor value的高低水平,可以将样本分为低因子水平、中因子水平和高因子水平三个cluster,可结合指标数据和分类菌群的功能进行重点研究探索。
2.6 缺失值估计
另外,对于样本中某些特征数据有缺失的情况,MOFA软件可以依据数据内部关系进行缺失值的估算,估算后的数据可进行热图的绘制。
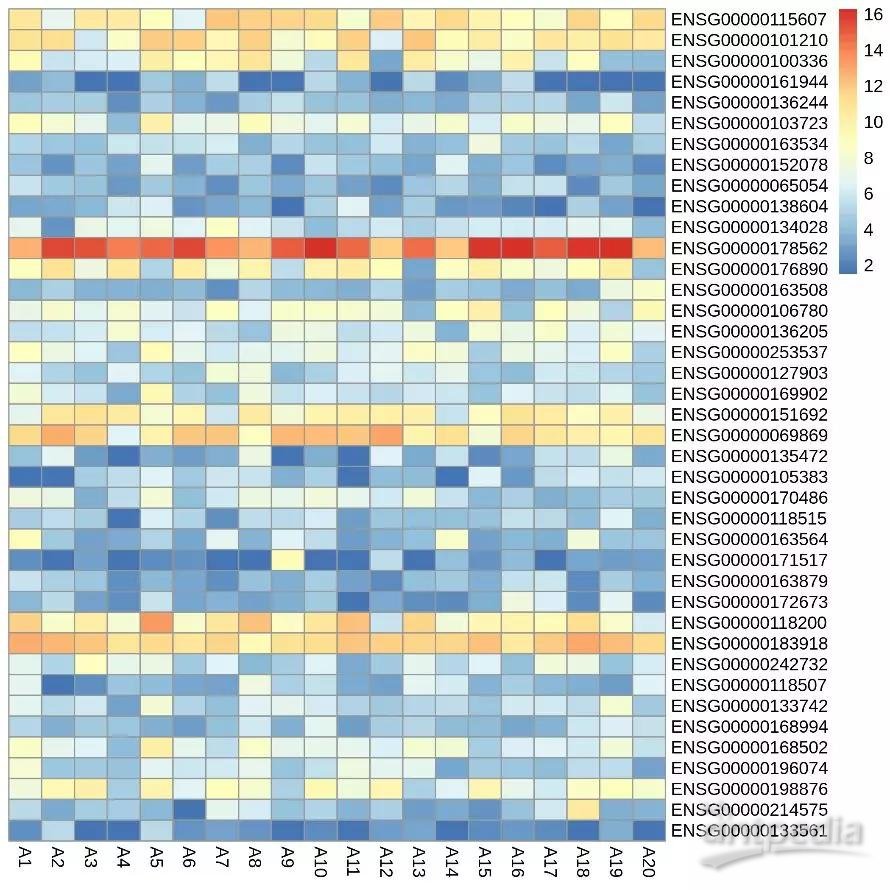
2.6.1 缺失值估算后的数据热图
其中每列代表一个样本,每行代表一个基因。上图表示填补缺失值后,转录组数据中样本和基因表达量的关系,从蓝色到黄色渐变代表表达量逐渐升高。
看到这里相信老师们对多组学因子分析软件MOFA有了一定的了解,以上分析,我们都有相关的案例和分析经验,感兴趣的老师欢迎和当地销售联系垂询~
参考文献
Ricard Argelaguet, Britta Velten, Damien Arnol et al. Multi-Omics Factor Analysis: a framework for unsupervised integration of multi‐omics data sets[J].Mol Syst Biol. 2018, 14:e8124.
END





















 首页
首页
