苏黎世联邦理工学院的Ruedi Aebersold团队一直致力于蛋白质组学的研究,其中包括开发将蛋白质组作为一个完整实体加以研究的相关技术。通过与SCIEX合作,该团队开发了SWATH® 质谱采集技术,这是一种能够同时碎裂多种多肽的数据非依赖性采集(DIA) 方法。得到的全面数据集可以通过回溯进行再挖掘,从而让研究获得尽可能多的信息。

Ruedi Aebersold教授,
苏黎世联邦理工学院生物系负责人
Ruedi Aebersold团队隶属于瑞士苏黎世联邦理工学院生物系,他们研究的重点是快速发展的蛋白质组学领域。该团队的一大研究重点是持续开发新的蛋白质组分析技术,其中质谱是他们首选的分析技术。生物系负责人Ruedi Aebersold教授解释说:“基本上,研究蛋白的方式有两种:基于亲和试剂的蛋白质组学方法以及质谱法。基于抗体的试剂很好用;但是,除了人类蛋白质组之外,抗体来源并不丰富;实际上,目前还没有可用于酵母、老鼠或苍蝇等物种的系统性抗体库。对于这些物种以及人类蛋白质组学而言,质谱就是首选方法。在目前可用于分析蛋白质和蛋白质组的工具中,质谱法是最通用、最客观公正的一种工具。它可以从头测序鉴定蛋白质,还能在一定程度上对翻译后修饰进行分析。我们甚至还能通过质谱了解蛋白质是如何折叠和相互作用的。”
他接着说道:“我们使用质谱已经有很长时间了,这些年来这项技术有了很大的进步。现在,质谱仪器的灵敏度已大幅提高,分辨率和扫描速度也更高、更快。因此,蛋白质的分析方法也发生了不小的变化。其中的一个重要里程碑就是定量技术的发展,这些技术可以确定蛋白质的丰度;另一个里程碑是软件的进步,先进的统计工具可以指出结果的对错,例如蛋白质的鉴定是否正确。第三项重大进步则是从数据依赖性采集或“鸟枪法”测序技术,转变到数据非依赖性采集的 SWATH 技术。这种转变目前仍在进行中,但我坚信 SWATH-MS的应用将推动蛋白质组学的变革,尤其是大规模蛋白质组研究项目的变革。”
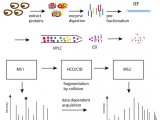
“数据依赖性采集的不足之处在于,重复分析时会得到不同的结果,这完全是因为在特别复杂的样品中,质谱仪器每次分析的可能是大量多肽的不同部分。SWATH大大缓解了这一问题;它可以同时碎裂多种多肽,而不是一次只碎裂一种多肽。这是技术进步的一个关键点,因为这样可以保证不会再有任何多肽被遗漏,也可以消除随机性。此外,数据分析也是一大挑战,因为这种采集方法会生成由来自不同母离子的碎片离子构成的复杂谱图,传统的搜索引擎无法对这种谱图进行有效分析。其实数据非依赖性采集 (DIA)并不是一个新概念,但最初没能推广开来,因为当时的质谱在进行数据采集时速度过慢或是表现不佳,而且复杂碎片离子谱图的评估问题总体上还未能得到解决。但是,当我看到SCIEX TripleTOF®5600+,我立刻清楚地意识到这台仪器不仅能执行DIA,还能解决数据采集问题。另外,数据分析的难题可以通过引入靶向抽提的策略进行根除。于是,我们与SCIEX 展开合作,一同实施SWATH技术并为其申请了ZL,现在这项技术已被广泛采用。”
“[SWATH] 有一个很大的优点,可以让我们从数据中最大程度地获取相关信息……”
“SWATH是蛋白质组学的理想之选,我们可以利用这项技术开展研究,例如,基因组的变异性是如何转化为各种表型的;另外,我们还可以利用这项技术开展生物标记物的研究。历史上第一次,我们可以对成百上千的相似样品进行可靠且适度的定量研究,并对它们进行真正有意义的比较,这些样品可以是类型完全相同但处于不同状态的细胞、从大量个体采集到的样品,或是从一个个体的不同组织中采集到的样品。由于样品众多,我们必须在样品准备阶段将实验误差降至最低,为此我们采用了压力循环技术 (PCT)。PCT是一种非常快速的样品准备方法,它可以规避很多固有的操作误差,从而获得可重复的多肽样品。凭借这项技术,我们还可以并行运行最多 96 个样品,这真的让我们受益匪浅。”
“对于参考数据的访问非常关键,尤其是在实施高度平行的靶向数据分析策略时。我们与 SCIEX 密切合作,一同构建了一个约由 11,000 种人体蛋白质构成的公共谱库,还完善了酵母和结核分枝杆菌蛋白质组的谱库,以及一些与老鼠相关的数据。一些实验室喜欢针对他们感兴趣的特定领域创建本地谱库;其他实验室则会使用DIA-Umpire,这个工具可以在分析SWATH数据,或是通常所说的DIA 数据的同时构建内部谱库。近期,有人将DIA-Umpire与其他四种已发表的蛋白质组工具进行了比较,其中包括我们自己的OpenSWATH 和SCIEX的PeakView™;使用不同的工具分析相同的样品数据,产生了精确的可比对结果。我们还开发了TRIC,这是一个用于靶向蛋白质组学数据处理的比对工具,特别适用于大样本量的数据;它会使用树构建算法非常精确地校准不同样品中的多肽,从而提高重现性。”
Ruedi Aebersold总结道:“ 关于SWATH,有一点特别有意思。它可以对采集的数据进行再挖掘,以寻找原先未曾预想过的蛋白质序列变异或修饰。例如,在通过肿瘤活检采集到数据集后,新公布的数据可能会表明存在特定的融合或基因组重排现象。SWATH允许我们回头查看原始数据并进行再挖掘,以确定是否存在或缺失报告的产物。这是一个很大的优点,可以让我们从数据中最大限度地获取相关信息;在分析珍贵的和无法再次获取的样品时,比如组织活检,这一点尤为重要。”





















































 首页
首页


