上周,《最强大脑》第五季强势回归!本季《最强大脑》以“致青春”为主旋律,寻找14~26岁的天才少年。都说“好看的皮囊千篇一律,有趣的灵魂万里挑一。”一群超高智商的少年,在这个冬季尽情的释放他们的活力。
选手们需要在紧张的比赛氛围中克服负面的心理情绪,高速运转大脑,极致细心,才能在百人之中脱颖而出。极大地考验选手们的耐力,抗压能力。小编看着也是紧张到手心出汗。

而在进行荧光定量 PCR Assay 的实验中,我们也接受到来自各方面的考验,需要我们克服重重困难,最终才能取得实验成功。下面小编将为你介绍荧光定量 PCR Assay 实验设计,在实验中助你一臂之力!
▼
聚合酶链式反应 (PCR)是分子生物学领域功能最强大的技术之一。采用 PCR 技术,利用序列特异性寡核苷酸、热稳定性 DNA 聚合酶和热循环,可将 DNA 或 cDNA 模板内的特异性序列拷贝或“扩增”数千至数百万倍。
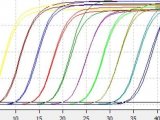
在过去的数年中,实时荧光定量 PCR 已成为 DNA 或 RNA 检测和定量的主要工具。采用上述技术,您可以实现精确的检测,其准确度低至2倍范围,起始材料的动态范围达6 至 8 个数量级。
成功的实时荧光定量 PCR Assay 设计和开发是获取准确数据的基础。前期规划将有助于管理在此过程中出现的实验差异。
第一步:
在着手实验设计前,应清晰地了解 Assay 的目标,尤其是需要回答哪些生物学问题。例如,旨在确定特定疾病状态下某个基因的相对表达水平的实验与旨在确定该疾病状态下病毒拷贝数的实验具有很大的差异。
第二步:
在确定实验目标后,识别适当的实时荧光定量 PCR 对照和进行实验优化的可能性。本部分将介绍实时荧光定量 PCR Assay 设计和执行的各个阶段。我们将从下面几个方面鉴别差异的来源、其在数据准确性方面的作用以及优化指导原则:
靶向扩增片段和引物设计
核酸纯化
逆转录
质控品和标准化
效率、灵敏度和可重复性标准曲线评估
实时荧光定量 PCR Assay 类型
基因表达谱分析是实时荧光定量 PCR 的常见应用之一,其通过评估转录本的相对丰度,确定不同样本的基因表达图谱。RNA 质量、逆转录效率、实时荧光定量 PCR 效率、定量方法及标准品基因的选择在基因表达实验中发挥了尤为重要的作用。
病毒滴度测定 Assay 的设计较为复杂。研究人员通常想要定量样本中的病毒拷贝数。一般采用已知基因组等效物或从已滴定的病毒对照品中采集的核酸生成标准曲线,通过与标准曲线进行比较实现滴度测定。实验成功与否取决于生成标准曲线所用的材料的准确度。根据靶点的性质——RNA 或 DNA 病毒——逆转录和实时荧光定量 PCR 的效率亦具有重要的作用。病毒是否为功能性病毒或该研究是否对总病毒颗粒进行分析,都将影响到 Assay 设计。
在拷贝数变异分析中,基因组进行重复或缺失分析。Assay设计,特别是标准曲线的生成将取决于是要求相对还是绝对定量。Assay 设计主要关注区分单拷贝差异需要的实时荧光定量 PCR 效率和准确度。
最后,等位基因分型 Assay 可以检测低至单核苷酸水平的差异。与上述方法不同,它需要检测终点荧光,确定 SNP 基因型。引物和探针设计是确保等位基因特异的交叉反应性低发生率的关键。
扩增片段和引物设计的考虑因素
靶向扩增片段大小、GC 含量、位置和 特异性
反应效率对实时荧光定量 PCR 数据的准确度极为重要,本指南将在后面的部分对此进行更详细的介绍。在理想状态下,每个循环后,PCR 反应中的每个靶点拷贝将被复制,全长靶分子数目倍增:这对应 100% 扩增效率。随着热循环过程的进行,效率的差异将被放大。因此,100% 效率的任何偏差均可导致潜在的错误数据。
使效率偏差最小化的一种方法是扩增相对较短的靶点。在特定的循环中扩增 100 bp 的区域相比扩增 1,200 bp 的靶点,更有可能实现完整合成。因此,实时荧光定量 PCR 靶点的长度一般为 60–200 bp。此外,较短的扩增片段受靶点-模板完整性差异的影响更小。如果核酸样本出现轻微降解,而靶序列较长,则上游和下游引物在同样的 DNA 片段中寻找其互补序列的难度将更大。
扩增片段的 GC 含量和二级结构是引起数据不准确的另一个因素。如果二级结构阻碍了 DNA 聚合酶的路径,则每次循环更有可能出现不完美的靶点扩增。理论上,设计的引物应可与中等 (50%) GC 含量且无明显 GC 延伸段的区域退火结合并扩增。对于 cDNA 扩增,最好使扩增片段位于转录本的 3’ 端附近。如果 RNA 二级结构阻止了某些转录本的全长 cDNA 合成,则这些扩增片段受影响的可能性更小 (图 1)。
图 1. 具有高度二级结构的 RNA 分子。
靶点特异性是影响数据准确度的另一个重要因素。设计实时荧光定量 PCR 引物时,应检查引物以确保其结合位点是基因组中独一无二的。这降低了引物扩增样本基因组中的其它类似序列的几率。引物设计软件程序自动去除位于起始基因组和掩蔽同源区域的靶序列,避免了这些位点的引物设计。
基因组 DNA、假基因和等位基因变异体
在检测基因表达水平时,需要关注 RNA 样本中的基因组DNA 残留。gDNA 可与目的靶点转录本共扩增,从而产生无效数据。通过设置不含有逆转录酶的对照反应 (RT 质控品) 检测基因组 DNA 污染;如果 RT质控品的 Ct 值高于稀释度最高的靶点生成的 Ct 值,则表示无 gDNA信号生成。但由于 gDNA 可竞争反应组分 (如 dNTP 和引物),因此会影响反应的效率。
实时荧光定量 PCR 中避免 gDNA 干扰的最佳方法是利用gDNA 中存在而 mRNA 中不存在的内含子,进行周到的引物 (或引物/探针) 设计。设计 TaqMan® Gene Expression Assay 时应尽可能使 TaqMan® 探针覆盖相邻两个外显子交界处。设计用于基于 SYBR® Green 染料检测的引物组时,应使引物与相邻外显子序列退火结合,或者使其中一个引物覆盖相邻两个外显子交界处。
当上游和下游 PCR 引物与同一个外显子退火结合时,则它们既可以扩增 DNA 靶点,也可以扩增 RNA 靶点。相反,当引物与相邻外显子序列退火结合时,大多数情况下仅可以扩增 cDNA,这是由于 gDNA 扩增片段包括内含子序列,从而导致扩增片段过长,无法在实时荧光定量 PCR 条件下实现高效扩增。
假基因或沉默基因是设计引物时也需要考虑的转录本变异体。它们是现有基因的衍生物,由于启动子或基因本身的突变和/或重排导致了功能丧失。引物设计软件程序可进行 BLAST 搜索,避免出现假基因及其 mRNA 产物。
等位基因变异体是一个基因的两种或多种独特的形式,它们具有相同的染色体位点。源自上述变异体的转录本相差一个或多个突变。设计引物时应考虑等位基因变异体,具体取决于研究的是一个还是多个变异体。此外,变异体之间的 GC 含量差异可改变扩增效率,并在熔解曲线上生成单独的峰,易被错误地诊断为脱靶扩增。设计引物时还应考虑可变剪接的变异体。
引物和探针的特异性、二聚体形成和自折叠
引物二聚体形成的最常见原因是正向和反向引物的相互作用,但也可能是由于正向引物与正向引物或反向引物与反向引物之间的退火或者单个引物自身的折叠引起的。在更复杂的反应 (如多重实时荧光定量 PCR) 中,引物二聚体更值得关注。如果以交错排列的方式形成二聚体 (通常是这种情况),则可出现一定的延伸,形成大小接近目的扩增片段的产物,且丰度随着循环的进行而增高。
一般而言,PCR 反应的起始靶点量越少,形成引物二聚体的可能性越大。上述潜在问题的积极方面在于,引物二聚体之间的相互作用弱于目的引物与模板之间的相互作用,此外,有很多方法可以最大程度地减少或消除此现象。

引物二聚体的主要问题在于它们可引起假阳性结果,特别是使用 DNA 结合染料 (如SYBR® Green I 染料) 的反应。另一个问题是它会导致与反应组分的竞争作用,使反应效率超出 90-110% 的理想范围。最后一个主要问题也与效率有关,是反应的动态范围可能缩小,从而影响反应的灵敏度。即便信号并非来自引物二聚体本身 (如 TaqMan®Assay),反应的效率和动态范围仍将受到影响。
有几款免费的软件程序可分析您的实时荧 光 定 量 PCR 引物设计,并 确 定 它 们 是 否 容 易 形 成二聚体或自折叠。AutoDimer 程序 (由美国国家标准与技术研究院的P.M. Vallone 编制) 是一款可同时分析各种引物的生物信息学工具 (图 2)。这尤其适用于多重反应领域。但虽然引物序列的生物信息学分析会大大降低二聚体形成的风险,但仍需要通过实验对二聚体形成进行监测。
图 2. AutoDimer 软件屏幕截图。 该软件可用于分析引物序列并报告引物内可能的二级结构区域 (可导致引物自折叠) 或可使引物相互退火结合的延伸段序列。
传统的引物二聚体筛查方法是凝胶电泳,二聚体会在凝胶的底部形成扩散的模糊条带 (图 3)。凝胶验证的问题之一是它的灵敏度不高,因此可能无法得出结果。但凝胶分析适用于验证从熔解/解离曲线获取的数据,这是检测引物二聚体的最佳方法。
图 3. 通过琼脂糖凝胶分析研究引物二聚体的形成。在热循环反应前,将核酸样本连续梯度稀释后加入 PCR 混合物中,然后从每种混合物中取相同体积上样,进行琼脂糖凝胶电泳。引物二聚体在凝胶的底部形成扩散条带。
采用 DNA 结合染料进行检测的实时荧光定量 PCR 运行结束后,应生成熔解或解离曲线。简言之,仪器从低温状态开始升温,低温下 DNA 呈双链且荧光强度较高,达到高温时,DNA 开始变性,荧光强度开始下降。在 PCR 过程中生成的各产物的 Tm 值处将观察到荧光出现明显下降。比较无模板对照 (NTC) 的熔解曲线峰与靶点峰,确定反应中是否存在引物二聚体。
理论上,包含模板的每个反应应观察到一个单峰,而 NTC 中应无荧光峰存在。在较低熔解温度下出现的比目的扩增片段更小、更宽的峰,且同时出现在 NTC 反应中的,通常是二聚体。同样,产物凝胶电泳常可用于验证熔解峰对应的产物的大小。
有些情况下,引物二聚体存在并不会影响实时荧光定量PCR 分析的总体准确度。一个常见的现象是引物二聚体存在于无模板对照中,但在包含模板 DNA 的反应中没有。这并不奇怪,因为在模板不存在时,引物更有可能相互作用。当有模板存在时,不利于引物二聚体形成。只要在NTC 中观察到的峰未出现在模板解离曲线上,引物二聚体就没有问题。
引物二聚体是一大类非特异性 PCR 产物中的一部分,包括当引物与不理想位点不完美匹配后退火结合形成的扩增片段。非特异性产物扩增值得关注是因为它们可发出荧光,人为地改变反应的 Ct 值。它们可以通过竞争反应组分影响反应效率,降低动态范围和数据准确度。在绝对定量分析中,需要报告精确的拷贝数,因此非特异性产物更值得关注。
标准凝胶电泳通常是实时荧光定量 PCR 特异性分析的第一步。它可以鉴别与您的靶点扩增片段大小不同的产物,但其条带会掩盖大小相似的扩增片段,且灵敏度有限。鉴于熔解曲线分析的准确度和灵敏度,它是确认凝胶电泳引物特异性评估的最值得信赖的方法。
尽管非特异性扩增应始终尽量避免,但在某些情况下,这些二级产物的存在并不会引起严重的问题。例如,如果以GC 含量不同的异构体或多个等位基因为靶点,则预计会出现多个产物。
引物设计的考虑因素
设计实时荧光定量 PCR 引物时有下列推荐:Primer Express®,OligoPerfect™ Designer 和 Vector NTI® 软件。请注意,引物设计软件程序,如我们的在线 OligoPerfect™ 设计程序和 Vector NTI® 软件序列分析软件,可与我们的在线订购系统无缝连接,因此您无需复制粘贴序列。这些程序可自动设计针对特定基因或靶序列的引物,其算法采用了下列指导原则,此外还可以进行已知序列同源物的全基因组 BLAST 搜索。
设计引物的长度一般为 18–28 个核苷酸
避免重复核苷酸延伸段
目标为 50% GC 含量,有助于防止错配稳定化
选择 Tm 值匹配的引物 (在 5°C 范围内)
避免一个分析中采用的所有引物之间以及各引物内部出现序列互补
本文部分图片来自网络,如因版权等有疑问,请在本文刊发30日内联系本账号。


















 首页
首页




