分享至
分享至



经费少、时间紧还想做性状定位,怎么办?
提到性状定位,我们通常会想到大群体、复杂算法、耗资巨大,例如全基因组关联分析。今天向大家介绍的BSA则不同,这种方法不仅材料容易准备、算法原理清晰明了,更重要的是不需要花费高额经费,十分适合性状初定位。
BSA混池分组分析法简介
BSA(bulked segregant analysis)混池分组分析法,根据目标性状表型对分离群体中的个体分别进行分组混合,依据目标性状的相对差异选择表型极端的个体分成两组,然后将两组的个体或株系DNA混合,形成相对的DNA混合池。通过在亲本和子代混池之间的多态性标记筛选即可完成对目标性状的定位。
BSA适用群体
关于性状定位常用的群体类型有两种,一种是自然群体(个体间遗传背景差异较大,通常来自不同地域、不同品种、甚至于不同进化程度的材料),另一种是家系群体(群体遗传背景较为相似,通常全部来自于两个亲本个体,在目标性状上有明显表型分离)。
BSA采用的是家系群体,常用的家系群体有F2、BC(n)、RIL、DH等等。对于家系群体的构建,亲本选择建议:同一物种前提下,目标性状尽可能差异大的两个个体。
BSA原理及分析逻辑
BSA中通常需要测序4个样本
两个亲本+两个子代混池
测序拿到原始数据后,可通过质控、与参考基因组比对、SNP calling等步骤得到各样本SNP标记分型信息。对于重测序而言,无论是BSA还是其他性状定位方法,SNP分型信息是后续分析的基础。

接下来如何从海量的SNP标记中筛选到与性状性格的那一部分,进而定位到性状相关基因是关键。对于“双亲健全”(因为实际情况可能只有两个子代混池)的BSA项目来说,是一个三步走策略:
Step 1. 筛选两亲本间纯合且差异的标记
家系群体构建时我们知道,首先选择在目标性状上差异明显的个体为亲本,表型有差异,跟表型相关的SNP标记就一定有差异。在亲本之间无差异的SNP可以先筛掉,剩余的亲本间纯合且差异的这部分再借助子代混池进一步聚焦。
Step 2. 子代混池筛选SNP标记并获得候选区间
标记与性状进行关联是性状定位最核心的部分,对于BSA而言,有两种关联方法,一种是ΔSNP-Index,一种是欧式距离(Euclidean distance, ED)。
ΔSNP-Index
SNP-Index可以理解成突变位点遗传物质来源于某一个亲本的频率,对于混池的某一个位点来说,SNP-Index即为该位点与某亲本序列相同的reads占总reads的比例。而Δ SNP-Index即为2个混池之间SNP-Index的差值。滑窗方式计算各区域的Δ SNP-Index,通过置信区间的设置可以判断区域是否与目的性状存在关联。

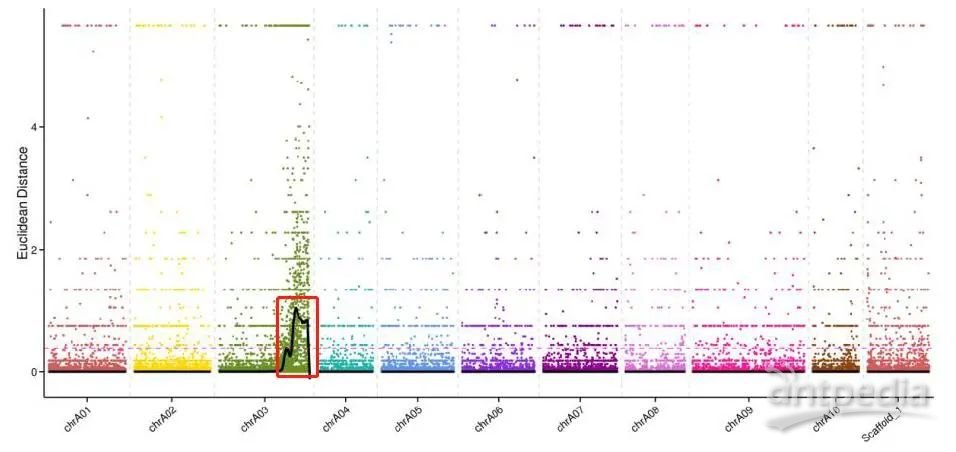
欧式距离(Euclidean distance, ED)
欧式距离计算将混池中每一个位点的突变信息抽象成坐标上的一个点,并计算混池之间每个相同位点之间的欧氏距离:
ED2=(A1-A2)2+(T1-T2)2+(C1-C2)2+(G1-G2)2
An、Tn、Cn、Gn分别指ATCG四种碱基在混池n中的频率
取所有位点拟合值的median+3SD作为分析的关联阈值,根据关联阈值判定候选区间。
Step 3. 区间内候选基因注释及功能富集
筛选SNP位点不是目的,关于结果,候选区域更具有生物学意义。确定候选区间后,注释区间内所包含的基因,并对候选基因集做KEGG、GO富集分析。
BSA方案设计
说到方案,主要涉及三个方面:亲本测序深度、子代混池样本数、子代混池测序深度。方案设计是否合理直接关系到结果定位的精度和项目费用。
极端混池的样本数量越多,混池测序深度越高,定位结果精度越高,但同时项目费用也会越高。有学者借助模式植物拟南芥专门针对子代混池样本数和测序深度对定位结果的影响进行了研究。结果发现,当混池样本数量达到20个,同时测试深度在20X以上的情况下,能够得到相对理想的定位结果。超过这个数量,随着成本消耗的增加,定位精度的提升效果有限。综合考虑,通常推荐方案如下:

亲本测序深度 | 10X |
子代混池样本数 | 20个 |
子代池测序深度 | 20X |
注:推荐方案不能适用于所有项目,具体方案要根据项目具体情况而定
BSA结果展示
高通量测序项目的结题报告内容向来比较多,包括各种形式的图表展示,抓住重点才能让我们对结果的解读准确而高效。对于BSA而言,要重点关注以下几个方面:
候选区域
候选区域内基因注释
基因功能富集
点击图片了解结果细节

< BSA-demo报告 >
BSA常见问题
Q1:没有参考基因组的物种可以做BSA吗?
BSA性状定位的方法适用于有参考基因组的物种。参考基因组组装质量好坏以及注释是否完全对BSA结果有一定的影响。建议尽量选择组装至染色体水平,且基因组注释较完全的参考基因组。
Q2:混池样本如何选择,一定要到20个吗?
混池的样本数通常选取极端性状(群体总样本数的5~10%内)的个体进行混池。混池中若极端性状个体数太少,可能造成样品数不够,不具备代表性;若样本数过高可能会引入杂合个体,产生干扰。需要根据群体的实际情况,首先保证表型足够极端,其次考虑样本数量。不一定必须到20。例如,某兔子毛色性状定位研究,通过两亲本杂交、F1个体混交,得到35只F2个体,将极端表型控制在20%以内,分别选择5只、6只进行混合,测序深度各为10X,也得到了较为理想的定位结果。
Q3:影响定位结果的因素?
评估定位结果主要参考候选区域大小和候选基因个数,影响它们的因素包括但不限于两亲本材料的差异大小、家系群体大小及性状分离情况、子代混池个数、亲本及子代混池测序深度、参考基因组水平、目标性状特点等。
Q4:子代混池构建是先提取在混合还是先混合再提取?
先提DNA再等量混合,可以减少系统误差,近年发表的多数文献都是先提DNA再等量混合。
Q5:BSA性状定位是否可以采用简化基因组测序?
简化基因组捕获的基因组区域有限,一般仅能捕获3%~30%,如果变异的区域正好没有捕获到就不能找到目标性状的基因了。因此用简化基因组的风险很大,不建议做简化基因组的BSA性状定位。
当然要做性状定位研究,除了BSA,还有遗传连锁图谱、全基因组关联分析等经典方案,有任何疑问可点击进入下方小程序提交需求,会有技术专家为您免费答疑解惑哦。