近期,Nature Methods 杂志技术编辑Vivien Marx发表文章 A dream of single-cell proteomics,探讨了单细胞蛋白组学的发展,提出了该技术有可能会面对的问题和潜在解决方案。

单细胞蛋白质组测序的梦想并不遥远(Credit: S. Larochelle, E. Dewalt, Springer Nature)
现在,很多实验室每年都在产生大量的单细胞基因组以及转录组的数据。但是在蛋白质组学研究中,高通量的方法尚未实现。蛋白质的研究工作比RNA或者DNA的研究要更难,比如说因为蛋白质比较黏、不可扩增并且容易降解等,但是科学家的前赴后继可能可以将单细胞mRNA与单细胞蛋白质组测量相整合。
20年前,Richard Smith和他的同事们首次对红细胞中的血红蛋白进行了表征【1】,虽然红细胞是个特例,因为其中主要组成成分就是血红蛋白,但这的确是单细胞分析。最近关于蛋白质组的研究方法,是由德克萨斯大学的Edward Marcotte与Eric Anslyn研究组开发的,能够以高度并行的方式产生氨基酸序列,该方法使用了Edman测序方式【2】,但是想要使用该方式进行单细胞的蛋白质组测序,还需要能够对细胞中低丰度的蛋白进行荧光标记。
想要实现单细胞蛋白质组测序,可能的几个思考方向是分离单细胞进行测序、样品制备技术进一步提高、多种技术联用、与物理学或者是生物信息学相联系等等。
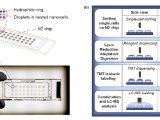
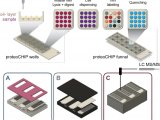
在单细胞样品制备方面,来自渥太华大学与牛津大学的团队使用单细胞计数(Single-cellmass cytometry)的方式去捕获造血作用中细胞命运决定以及追踪在13个时间点谱系中细胞转录因子表达谱的变化。而美国东北大学的Nikolai Slavov团队建立了质谱分析单细胞蛋白质组(Single cell proteomics by mass spectrometry ,SCoPE-MS)的方式(图1)【3】。Slavov团队重新考虑了样品制备过程中蛋白质损失的问题,并通过利用聚焦声波对裂解细胞进行超声处理进而发展了SCoPE-MS的方式,并且通过使用纯水进行循环,可以进一步降低该技术的使用价格。

图1 SCoPE-MS原理示意图
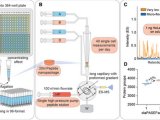
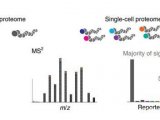
哈佛医学院,Peter Kharchenko研究组一直在开发用于单细胞的计算工具用于解决异构问题的RNA-seq数据分析(图2)。随着RNA-seq被应用于复杂的研究类型中,涉及到来自不同实验室的大量样本信息,计算工具开发的挑战变得越来越大。而这种计算工具也可以用于单细胞蛋白质组学。但是单细胞蛋白质组学的数据量会比RNA-seq所分析的数据更多,因此会对计算工具的要求以及计算模型的建立要求更高。

图2 在单细胞蛋白质组学中,量化和整合有关细胞行为、信号和调控网络的数据的工具
Ruedi Aebersold实验室在进行少量细胞的分析,利用8-10种荧光颜色的多参数荧光激活细胞分选(FACS)对细胞进行聚类,并根据相似性对细胞进行分组。他们不是先研究单个细胞而后对它们进行平均或组合,而是先将它们组合起来然后测量平均值。
更多的实验室现在可以使用大型数据库来预测蛋白质是如何相互作用的。除了测量蛋白质的丰度或浓度,癌症研究实验室还想定量地跟踪激酶的活性,了解有多少磷酸化位点以及有多少靶蛋白可以磷酸化。因为相比于蛋白质的丰度,蛋白质分子的活性更为重要。生物学领域越来越多的工具正在出现,可以量化细胞内和细胞间的信号传导和调控网络的不同方面,而单细胞的蛋白质组学有助于解决这个问题。
越来越多的实验室希望将mRNA与单细胞蛋白质组学的测量结果相联系,但是除了在转录和翻译过程中细胞与细胞之间的差异以及不同水平的蛋白质周转,还有许多影响细胞、mRNA和蛋白质的外部因素都会对mRNA与蛋白质之间的对应关系产生影响。
Aebersold和Jurg Bahler研究组通过观察细胞的两种状态:静息状态与快速增殖状态,对裂变酵母中的转录组和蛋白质组进行了量化【4】。该研究展示了很多令人惊讶的发现,揭示了基因调控、转录、蛋白质生产和生理学之间的关系。当分裂酵母菌进入休眠状态时,蛋白质和mRNA都会以特定的方式被下调。当调整到静止细胞中较低的细胞体积时,蛋白质的数量下降了增殖细胞水平的10%左右。mRNA水平下降,而蛋白质水平下降到更少。
Aebersold说,一个人类细胞包含数百个mRNA拷贝,其中蛋白质的数量有数万个。他说,人们可能在每个细胞中只发现少量低表达mRNA的拷贝,这使得确定一个细胞所处的时空特点变得越来越重要,而得出错误结论的风险再次浮出水面。从生物学的角度来解释单细胞数据有很多陷阱,技术很重要,但在开发和使用它们时,必须同时注意它们的好处与缺点。
众多实验室前赴后继地采用了一系列不同的方法进行研究,多种不同进展的结合将使得我们越来越接近单细胞蛋白质组学这一梦想的实现。我们需要从细胞中有效地提取蛋白质、通过特定的表面进行富集、样品需求越来越少高敏感性质谱、增强识别灵敏度的标记技巧以及在质谱数据采集和计算处理方面的进展。尽管如此,最近的一些进展包括新的大规模数据采集方法和更多的已知光谱库,大大增加了挖掘数据的可能性。在技术开发的早期阶段,我们有多种多样的方式来获得信息。鉴于这一点,我们不能将所有的“鸡蛋放在同一个篮子里”。
原文链接:
https://doi.org/10.1038/s41592-019-0540-6
参考文献
1 Hofstadler,S. A., Swanek, F. D., Gale, D. C., Ewing, A. G. & Smith, R. D. Capillaryelectrophoresis-electrospray ionization Fourier transform ion cyclotronresonance mass spectrometry for direct analysis of cellular proteins. Analytical chemistry 67, 1477-1480, doi:10.1021/ac00104a028(1995).
2 Swaminathan,J. et al. Highly parallelsingle-molecule identification of proteins in zeptomole-scale mixtures. Nature biotechnology,doi:10.1038/nbt.4278 (2018).
3 Budnik,B., Levy, E., Harmange, G. & Slavov, N. SCoPE-MS: mass spectrometry ofsingle mammalian cells quantifies proteome heterogeneity during celldifferentiation. Genome biology 19, 161, doi:10.1186/s13059-018-1547-5(2018).
4 Liu,Y., Beyer, A. & Aebersold, R. On the Dependency of Cellular Protein Levelson mRNA Abundance. Cell 165, 535-550,doi:10.1016/j.cell.2016.03.014 (2016).






















 首页
首页


