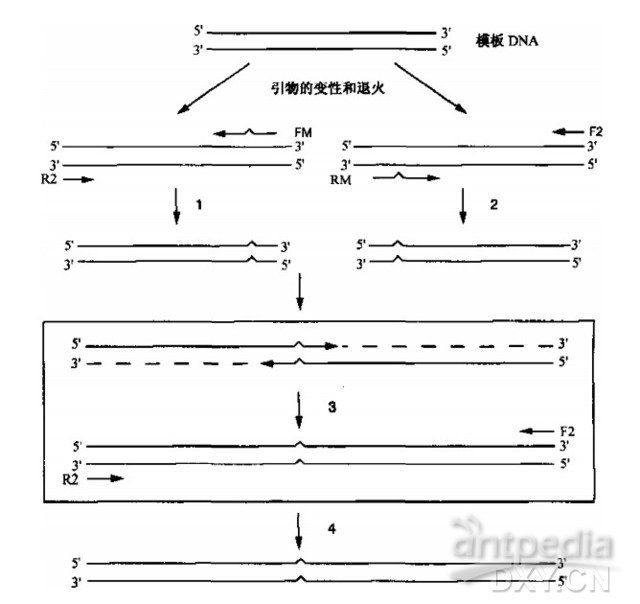
重叠延伸法进行特异位点诱变需要四种引物(请见图 13-4)(Higuchi et al.1988, Hetal.1989)。本实验来源于分子克隆实验指南(第三版)下册,作者:〔美〕J. 萨姆布鲁克 D.W. 拉塞尔。
| 试剂、试剂盒 | 扩增缓冲液热稳定 DNA 聚合酶琼脂糖凝胶或聚丙烯酰胺凝胶寡核苷酸引物模板 DNA |
|---|
| 仪器、耗材 | 带屏障装置的自动移液器吸头微型离心管酶可调式移液器可按所需扩增条件设定程序的热循环仪 |
|---|
| 实验步骤 | 材料

缓冲液和溶液
贮存液,缓冲液的成分及所需试剂请见附录 1。
将贮存液稀释释到适当的浓度。
10x 扩增缓冲液
含有四种 dNTP 的混合溶液,每种 dNTP 浓度为 20 mmol/L。
酶和缓冲液
热稳定 DNA 聚合酶
为了避免碱基的错误掺入,在重叠延伸诱变反应中应使用带 3'—5'外切核酸酶校对能力的髙效热稳定 DNA 聚合酶。另外. 使用的 DNA 聚合酶一定不能具有催化非模板性掺入腺苷酸残基的能力。具备此种特性的 DNA 聚合酶包括 Puo DNA 聚合酶 (Boehringer Mannheim),rTth DNA 聚合酶 XL(Pwkin Elmer)。VentR DNA 聚合酶(New England Biolabs) 和 Pfu DNA 聚合酶 (Stmagene)。
为长片段 PCR 反应所设计的热稳定 DNA 聚合酶混合物也适用于重叠延伸诱变反应。
凝胶
含 0.5ug/ml 溴化乙锭的琼脂糖凝胶(1%) 或聚丙烯酰胺凝胶
请见步骤 6 和 11。
核酸和核苷酸
寡核苷酸引物
每种引物的长度应为 20~30 个核苷酸. 含有大致相同数量的四种碱基,G 和 C 残基的分布应均匀,较低的形成稳定二级结构倾向。诱变引物 FM 和 RM 之间至少有 15 个碱基的重叠序列(请见图 13-4), 错配碱基应位于引物序列的中部。在扩增 DNA 片段的 3'和 5'端引物(即具有野生型序列 R2 和 F2 引物)可以设置单一的限制性酶切位点,以便于后续步骤中诱变 DNA 片段的克隆。引物设计的一般原则,请见第 10 章中导言部分关于核苷酸引物的设计。
在 DNA 自动合成仪上合成的寡核苷酸引物一般不需要纯化,可直接用在重叠诱变中。
模板 DNA
诱变中使用的模板通常是含感兴趣的基因或 cDNA 的质粒 DNA。将 DNA 以 1ug/ml 的浓度溶解在含低浓度 EDTA(<0.1 mmol/L) 的 10mmol/L Tris-Cl(pH7.6) 的溶液中。
专用设备
带屏障装置的自动移液器吸头
微型离心管(0.5 ml 扩增用薄壁管)
可调式移液器
可按所需扩增条件设定程序的热循环仪
如果热循环仪不配备加热盖装置,使用矿物油或石蜡油以防止 PCR 过程中反应混合物液体的挥发。
其他试剂
本方案步骤 11 需要的试剂列在第 1 章方案 17 或 19 中。
本方案步骤 12 需要的试剂列在第 12 章方案 3,4 或 5 中。
方法
1. 依据材料和方法中的要求和已知 DNA 序列设计合成寡核苷酸引物 FM,RM,R2 和 F2。
2. 在一个 0.5 ml 的微量离心管或扩增管中混合以下试剂,组成 PCR 反应 1。
模板 DNA 约 100ng
10x 扩增缓冲液 10ul
20 mmol/L4 种 dNTP 混合溶液 1.0ul
5umol/L 引物 FM(30pmoles) 6.0ul
5umol/L 引物 R2(30pmoles) 6.0ul
热稳定 DMA 聚合酶 1~2 单位
加水至 100ul
3. 在第二个微量离心管或扩增管中混合以下试剂,组成 PCR 反应 2。
模板 DNA 约 100ng
10x 扩增缓冲液 10ul
20 mmol/L4 种 dNTP 混合溶液 1.0ul
5umol/L 引物 RM(30pmoles) 6.0ul
5umol/L 引物 F2(30pmoles) 6.0ul
热稳定 DNA 聚合酶 1~2 单位
加水至 100ul
4. 如果热循环仪没有加热盖,用一滴石蜡油(约 50ul) 覆盖 PCR 反应液。将反应管置于热循环仪中。
5. 用下表中给出的变性、退火、聚合所需的时间和温度扩增 DNA 片段。
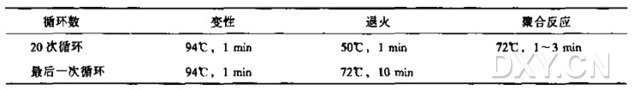
上述反应条件适合于 0.5 ml 薄壁管和 100ul 反应体积,以及 Perkin-Elmer 9600、9700,Master Cycler(Eppendorf) 或 PTC 100(MJ Research) 热循环仪。使用其他类型的仪器或不同的反应体积时需调整上述反应条件。
聚合反应所需的时间应通过计算热稳定 DNA 聚合酶的聚合效率及 DNA 模板长度来获得。
应根据诱变引物序列调节退火反应的溫度。
6. 各取以上两组 PCR 反应产物的 5% 进行琼脂糖或聚丙烯酰胺凝胶电泳估计扩增靶 DNA 的浓度。
7. 用第 5 章中描述的方法之一纯化以上两组 PCR 产物。对产物进行纯化往往增加步骤 8 中所希望的扩增产物的产量。并可降低错误扩增产物的背景。(选做)
8. 在一个灭菌的 0.5 ml 的微量离心管或扩增管中混合以下试剂进行扩增反应,连接靶基因的 5'和 3'端。
PCR1 扩增产物(步骤 2) 约 50ng
PCR2 扩增产物(步骤 3) 约 50ng
10x 扩增缓冲液 10ul
5umol/L 引物 F2(知 pmoles) 6.0ul
5umol/L 引物 R2(30pmoles) 6.0ul
热稳定 DNA 聚合酶 1~2 单位
加水至 100ul
9. 如果热循环仪没有加热盖,用一滴轻石蜡油(约 50ul) 覆盖 PCR 反应液。将反应管置于热循环仪中。
10. 用步骤 5 给出的变性、退火、聚合反应所需的时间和温度进行扩增。
11. 取 PCR 反应产物的 5% 进行琼脂糖或聚丙烯酰胺凝胶电泳估计扩增的靶 DNA 浓度。
如果引物 F2 和 R2 序列中加进了限制酶切位点,用这些限制酶消化扩增的 DNA, 然后把它们亚克隆至适当的栽体。也可以将步骤 10 扩增的 DNA 片段磷酸化,然后连接到用限制酶切割成钝端的载体中。
12. 克隆后确证扩增 DNA 片段的全序列,保证在操作过程中没有产生除引物 FM 和 RM 携带突变之外的其他突变。
所有 DNA 聚合酶在体外反应中都显示出可测量的错误掺入率。 |
|---|




 首页
首页


