
上期我们介绍了蛋白质组学的主要研究内容(蛋白质组学专题 | 一文读懂蛋白质组学研究策略及研究内容)包括蛋白质组定性、定量蛋白质组学、翻译后修饰蛋白质组以及蛋白质相互作用研究;同时介绍了基于自下而上(bottom-up)的基本研究策略。本期将给大家介绍当前蛋白质组学的主流技术。
在基于质谱的蛋白质组学方法学中,其核心方法主要包括蛋白质组定性和蛋白质组学定量两大类,其中定量蛋白质组学是蛋白质组学技术中的核心,当前zui主流的方法包括基于无标记定量(label-free quantification)[1]、基于等重同位素标记法定量(TMT/iTRAQ quantification)[2-4]以及基于质谱数据非依赖型采集模式的DIA/SWATH技术(如图1)[5, 6]。根据研究对象的不同,研究目标的不同,或使用仪器的不同,可衍生出多种蛋白质组学方法,但其基本方法离不开上述主流方法。

✦根据仪器不同
3D label-free,3D TMT, 3D DIA,4D label-free,4D DIA等
✦根据研究对象不同
血液蛋白质组、尿液蛋白质组、宏蛋白质组、微量蛋白质组、单细胞蛋白质组、空间蛋白质组......
✦根据研究目标不同
●翻译后修饰蛋白质组:磷酸化、乙酰化、糖基化、泛素化 ......
●外泌体蛋白质组
●亚细胞器蛋白质组:叶绿体蛋白质组学、线粒体蛋白质组学、细胞膜蛋白质组学…...
为了让童鞋们更好地理解不同方法的区别,小迈从各技术的实验步骤出发,对各种技术加以区分(图2)

■ ■ ■ ■ ■
图2 蛋白质组学各技术的实验步骤
●Label-free不分级:Step1→Step2→Step3→Step6→Step7→Step8
●Label-free 分级:Step1→Step2→Step3→Step5→Step6→Step7→Step8(不常用)
●Direct DIA:Step1→Step2→Step3→Step6→Step7→Step8(质谱采集模式为DIA)
●Library DIA:Step1→Step2→Step3→Step5→Step6→Step7→Step8(建库时质谱采集模式为DDA,其余为DIA)
●TMT/iTRAQ定量: Step1→Step2→Step3→Step4→Step5→Step6→Step7→Step8
●若为翻译后修饰,则需在上质谱前或肽段同位素标记前进行特定翻译后修饰肽段富集。
Label-free定量是最简单,也是最早使用的蛋白质组学定量方法。主要包含的实验步骤有:Step1→ Step2→Step3→(Step5)→Step6→Step7→Step8(图2)。在早期的文献中,由于质谱仪性能的限制,为了增加肽段/蛋白的鉴定深度,通常会在肽段层面先进行离线预分离(offlinefractionation,Step6)(图2),但增加离线分离也会增加定量误差。随着质谱仪性能的提升,现在很少有人使用这种策略。另外,label-free定量方法中质谱仪采用数据依赖型采集模式(DataDependent Acquisition,DDA)(关于质谱仪的基本原理及不同采集模式的介绍,小迈将在后续的推文中陆续上线)。
为了便于理解,我们以实际案例出发进行介绍。假设我们有Sample1和Sample2两个样本。使用label-free定量方法的基本策略为,将2个样本分别进行总蛋白提取、总蛋白酶解,此时得到2个样品的肽段,接着分别上质谱检测,得到2个质谱原始文件,这2个文件分别代表我们前面提到的2个样品,最后将这2个文件一起进行搜库、分析,得到最终定性、定量结果(图3)。

■ ■ ■ ■ ■
图3 label-free定量的基本策略
✦Label-free定量方法的优势
●实验操作简单,成本低
●不受样本数限制,样本数较大时无明显批次效应,因此适合大样本场景
●具有更大的线性动态范围
●基于timsTOF Pro2的4D label-free技术在检测深度上可以和传统TMT媲美
✦Label-free定量方法的劣势
●定量稳定性受实验操作者影响较大
●定量结果准确性略低于TMT
●一般质谱仪的检测深度不及TMT定量技术
●缺失值占比较高(和TMT、DIA相比)
TMT/iTRAQ定量属于等重同位素标记定量方法,是同位素标记定量方法中zui流行的一种,用的是同位素的原理,通过将不同的同位素标签与肽段特异氨基酸位点相连实现不同来源的肽段标记。该类试剂的化学结构由报告基团、平衡基团和反应基团三部分组成(图4),在特定组合的试剂盒内,其三种基团通过不同位置的C13、N15同位素组合保证总分子量恒定,而报告基团具有不同分子量,从而实现标记肽段后的肽段来源区分。其中iTRAQ试剂盒包含iTRAQ4-plex和iTRAQ8-plex两种,TMT试剂盒包括TMT6-plex, TMT 10-plex, TMT-11plex, TMT Pro 16-plex和TMTPro 18-plex。本文以TMT10-plex为例,介绍其基本原理和实验方法流程。

■ ■ ■ ■ ■
图4 TMT试剂的化学结构通式
TMT 10-plex的分子结构式与TMT6-plex完全相同(图4),分子式为H(20)C(8) 13C(4) N 15NO(2),只是13C和15N的数目和位置有所不同。表1为ETD和HCD两种碎裂模式下产生的报告基团的分子量。可以看到,TMT10-plex包含10种结构式完全相同但报告基团分子量不同的试剂。
表1 TMT10-plex报告基团分子量
标记试剂 | 报告基团分子量(HCD) | 报告基团分子量(ETD) |
126 | 126.127726 | 114.127725 |
127N | 127.124761 | 115.124760 |
127C | 127.131081 | 114.127725 |
128N | 128.128116 | 115.124760 |
128C | 128.134436 | 116.134433 |
129N | 129.131471 | 117.131468 |
129C | 129.137790 | 116.134433 |
130N | 130.134825 | 117.131468 |
130C | 130.141145 | 118.141141 |
131 | 131.138180 | 119.138176 |
其基本原理(或流程)为,将不同的试剂和不同的样本反应、标记,使特定样本中的所有肽段标记某一种特定标记试剂。所有样本标记完成后,取等量肽段混合,然后离线分离、上质谱检测。在质谱检测时,由于所有试剂的总分子量相同,来自不同样本的同种肽段的一级母离子表现为一个峰,相当于该肽段的多个样本的信号叠加,这样更有利于质谱选中打二级谱。当肽段母离子被选中并碎裂时,来自不同样本的标记试剂的报告基团便被碎裂出来并形成游离的离子,从而被质谱检测到最终实现定量(图5)。


■ ■ ■ ■ ■
图5 TMT标记定量的实验流程
优势:
实验引入的误差更少,定量结果更准确
一次同时可标记多达18个样本
肽段标记混合后实现离线分离,检测深度更高
劣势:
分析的样本数有限制,多批次标记存在批次效应
存在压缩效应,定量出的结果比实际结果偏低
无法进行有无区分
DIA的全称是DataIndependentAcquisition,可以看出,它是一种新型的质谱采集模式。我们知道,传统的质谱采集模式多为DDA,即数据依赖型采集模式,比如上文提到的label-free定量、TMT/iTRAQ定量。我们来简单理解一下,DDA是有选择性的对肽段母离子做二级碎裂(根据母离子强度又高到底依次选择10-30个),而DIA会选择全部肽段母离子做二级碎裂。因此,DIA可获取到更多的数据,而DDA会丢失掉绝大部分肽段信息(图6)。由于DIA采集模式可以将全部肽段母离子碎裂成更短的二级碎片离子,可以在碎片离子层面完整重现nanoLC的色谱峰图,因此DIA定量可以利用碎片离子随时间的积分来表征肽段含量,而利用二级碎片离子定量本身比一级离子定量结果(label-free)更准确,随机性误差更小。本质上,DIA也是一种label-free定量方法。
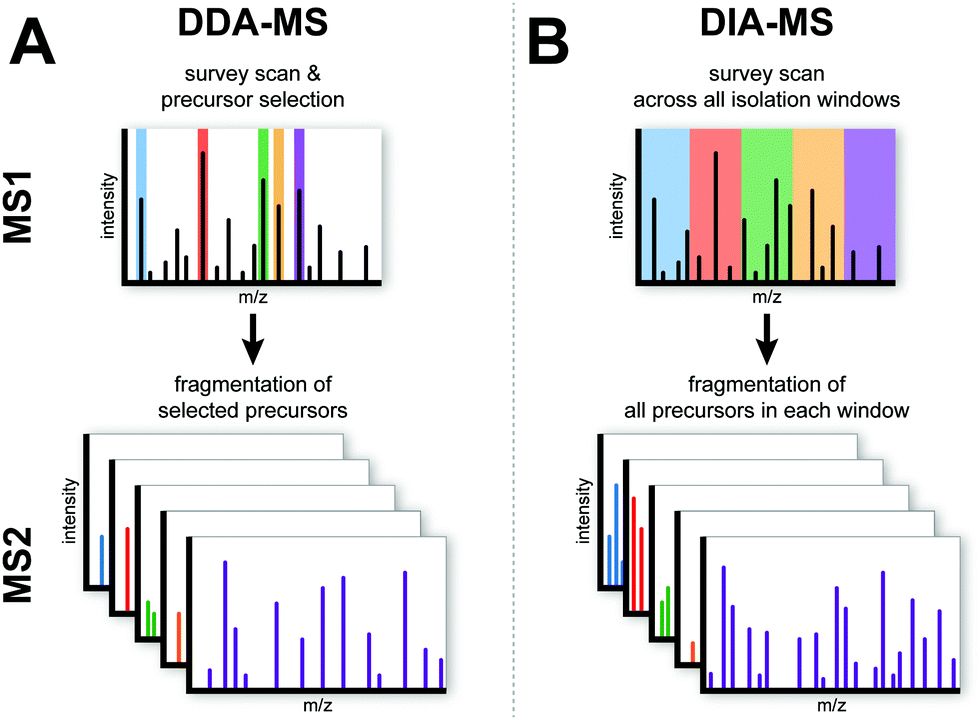
■ ■ ■ ■ ■
图6 DDA和DIA采集模式。图A为DDA采集模式,该模式下,从MS1全扫中选择强度较高的有限个母离子(窄窗口),并在后续扫描中逐个选中并进行二级碎裂;图B为DIA采集模式,将MS1的整个质荷比区域分成多个窗口(窗口),每个窗口包含多种肽段母离子,在后续的扫描中,一次扫描同时碎裂一个窗口中的全部肽段母离子。
上面提到,DIA采集模式会会按照质量窗口对所有肽段母离子做二级碎裂,而非按离子强度选择。其基本思路是:将一个MS1全扫描内的所有肽段母离子按质荷比(m/z)从小到大分割成多个质量窗口,紧接着,在后续的二级扫描中会逐步选择每个窗口中所有母离子并被送入碰撞室进行高能碰撞,形成二级碎片离子。由于采集的数据更多更全,反馈在最终的定量结果中,具有更少的缺失值,定量变异性更小;由于使用了MS2碎片离子信息定量,因此定量结果比label-free更准确。本质上,DIA也是一种label-free定量方法,因此,其前处理方法和策略跟传统的label-free完全相同。
DIA数据解析。DDA中一张二级谱图理论上仅为一种肽段母离子的碎片离子,而DIA中一张二级谱图理论上包含多种肽段母离子的碎片离子。因此,DIA数据比DDA更为复杂,数据解析时不能像DDA那样利用理论蛋白序列库和二级谱图比对。在DIA数据解析时,常常先使用DDA模式构建一个包含肽段序列、保留时间、碎片离子质荷比及强度信息的谱图库,通过谱图库中的碎片离子和DIA采集数据中的碎片离子的比对来识别DIA中某一张二级谱图中含有的肽段序列,从而完成肽段鉴定。再利用DIA采集模式的数据完整性的特点,对碎片离子构建提取的离子流色谱峰(XIC),并计算峰面积。接着根据碎片离子峰面积推断肽段峰面积,再根据肽段峰面积推断蛋白峰面积。值得一提的是,随着仪器的不断更新(尤其是基于离子淌度分离的timsTOFPro系列仪器的推出)以及深度学习等机器学习算法的应用,dia数据解析开始朝着不建库直接利用dia数据和蛋白序列完成肽段/蛋白的鉴定和定量。小迈后期专门抽出一期给大家科普一下。
引用文献
1. Neilson, K.A., et al., Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics, 2011. 11(4): p. 535-53.
2. Choe, L., et al., 8-plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer''s disease. Proteomics, 2007. 7(20): p. 3651-60.
3. Ross, P.L., et al., Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics, 2004. 3(12): p. 1154-69.
4. Thompson, A., et al., Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem, 2003. 75(8): p. 1895-904.
5. Gillet, L.C., et al., Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics, 2012. 11(6): p. O111 016717.
6. Sidoli, S., R. Fujiwara, and B.A. Garcia, Multiplexed data independent acquisition (MSX-DIA) applied by high resolution mass spectrometry improves quantification quality for the analysis of histone peptides. Proteomics, 2016. 16(15-16): p. 2095-105.


心动不如行动,
您只需提供样本,我们负责给您一幅满意的答卷!
有需求请询迈维代谢当地销售或者加微信metware888
99%的代谢组学研究者都在阅读下文:
●生信小工具 | 手把手教你从序列查找开始,绘制漂亮的进化树

客服微信:metware888
咨询电话:027-62433042
邮箱:support@metware.cn
网址:www.metware.cn
我就知道你“在看”
![]()

























 首页
首页
