
表达谱的异常与包括癌症在内的人类疾病息息相关,不仅适用于编码蛋白的信使RNA(mRNA),同时还适用于人类转录组中的非编码转录本。目前,人类基因组中75%以上的组分能够产生出相应的的转录产物,但其中仅有2%具备编码蛋白的功能。由此可见,不具备编码蛋白功能的“暗物质”,即非编码转录本,占很大比例。在非编码家族中,转运RNA(tRNAs)、小核RNA(snoRNA),以及微小RNA(microRNA,miRNA)等的相关研究已经较为成熟。但是,仍然有大量的非编码RNA(non-codingRNA,ncRNA),对其功能及作用机制并无深入了解。环状RNA(circularRNA,circRNA)是广泛且多样地存在于多种生物细胞中有调控基因表达作用的一类内源性ncRNA分子,具有结构稳定和组织特异性表达等特征。对circRNA的逐步认识不仅丰富了竞争性内源RNA(competingendogenousRNA,ceRNA)的调节网络,大量circRNA的发现及其结构和功能的阐明不仅可以使我们更加深入地了解疾病的发生机制,而且为相关疾病的预防诊断和治疗提供了新的方向。本文着眼的疾病为某疾病,通过整合多个数据库中不同类型的RNA(包括circRNA、miRNA和mRNA),分别筛选与正常组织比较下,肿瘤组织中表达水平显著改变的RNA,基于此构建ceRNA调控网络和研究疾病机制。
1、表达数据筛选
表1.检索得到的RNA表达谱列表。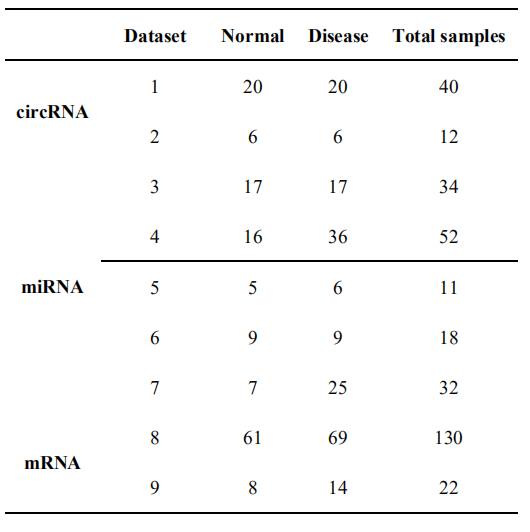
2、数据预处理
从NCBIGEO和EBIArrayExpress下载表1中所列编号的RNA表达谱数据,分成Affy和Agilent等原始格式为CEL和TXT的两种类型表达谱数据。对原始格式为TXT的表达谱文件,根据注释平台进行探针对应基因的注释,一个基因能对应到多个探针(多个表达值),我们对其求平均值,作为该基因对应的唯一的表达值。
我们利用R3.4.1中limma包Version3.32.5(对数据进行log2对数化,使基因表达数据由偏态分布转化为近似正态分布,然后用中位数标准化方法对数据进行归一化处理;对下载得到的原始CEL格式数据,利用R3.4.1语言中的oligoVersion1.41.1(http://www.bioconductor.org/packages/release/bioc/html/oligo.html)对数据进行原始CEL格式数据的转换,中位数法进行缺失值补充,背景矫正(MAS法)以及用分位数法(quantiles)进行数据标准化。
3、显著差异表达的筛选
①.显著差异表达的circRNA的筛选
对两个标准化后的circRNA表达谱数据,以每个数据集中的正常样本为对照,利用Limma包分别在两个数据集中筛选显著差异表达的circRNA,选取p值小于0.05且|logFC|>0.585(倍数值为1.5倍)作为是筛选差异的阈值,然后比较两个数据集中的显著差异表达circRNA,选取其交集且在差异方向上保持一致的circRNA作为综合两数据集的显著差异circRNA。
②.显著差异表达的miRNA和mRNA的筛选
由于miRNA和mRNA的数据集个数均超过了3个,因此采用R3.4.1语言中的MetaDEversion(https://cran.r-project.org/web/packages/languageR/index.html)进行一致性显著差异的筛选。Meta分析的主要目的是将多个研究成果综合反映出来,能够汇集多个实验数据集合,增强统计能力,筛选出更加可靠的基因。我们首先采用R3.4.1语言中MetaQC包Version0.1.13(https://cran.r-project.org/web/packages/MetaQC/index.html)的质量控制标准对数据集进行客观质量控制,结合主成分分析(principalcomponentanalysis,PCA)二维图及标准平均等级得分(standardizedmeanrank,SMR)共同评估和筛选数据集合。
MetaQC定量的质量控制标准:
1).内部质量控制(internalqualitycontrol;IQC),研究数据集合对象之间基因表达值结构同质性检验;
2).外部质量控制(externalqualitycontrol;EQC),与通路数据库表达结构一致性检验;
3).精度质量控制(accuracyqualitycontrol;AQCg),探测显著差异表达基因的精确度或者通路的识别(AQCp);
4).显著差异表达基因(consistencyqualitycontrol;CQCg)或者通路(CQCp)排序的一致性。通过MetaQC质控检验后得到的可靠数据集合将进一步采用MetaDE包中的MetaDE.ES筛选显著差异表达基因。我们阈值参数的选取:首先保证每个被筛选到的基因来源同质无偏倚,即在每个数据集中的表达情况一致,因此选取tau2=0,Qpval>0.05为同质检验参数;然后选取FDR<0.05作为基因组间表达差异显著性阈值。
4、显著差异表达RNA双向层次聚类
根据RNA表达在不同状态的同种生物中表达的特异性,相同的组织在不同疾病状态下表达是有显著的差别的。对筛选得到的显著差异的RNA,用R3.4.1语言中的pheatmap包(Version1.0.8)(https://cran.r-project.org/package=pheatmap)对表达值进行基于欧几里得距离的双方向层次聚类热图进行展示。
5、miRNA连接关系的预测
生物网络体现了基因之间或者基因与其他功能或通路之间的相互联系,通过网络分析可以发现基因影响生物体的协助脉络,并能在复杂的作用链条中,系统地挖掘基因发挥作用的真实线索。近年的研究表明基因的转录调控受到多种模式的影响,其中miRNA调控是重要的调控模式。现已发现,基因的转录后调控并不是简单的miRNA-mRNA的沉默机制,而是一个复杂的调控网络:很多非编码RNA分子富含microRNA结合位点,在细胞中起miRNA海绵(miRNAsponge)的作用,进而解除miRNA对其靶基因的调控作用,改变靶基因的表达水平,这一作用机制被称为竞争性内源RNA(ceRNA)机制。而在ceRNA机制中,circRNA是典型的miRNA海绵吸附体。因此,通过与miRNA相互作用,circRNA在疾病中发挥着重要的调控作用。我们通过以下几个步骤对mRNA、miRNA和circRNA之间的连接关系进行挖掘。
①.circRNA-miRNA的连接关系
我们利用starBaseVersion2.0数据库(http://starbase.sysu.edu.cn/index.php)中circRNA调控miRNA关系作用对,从中挑选出显著差异表达的circRNA与miRNA间的连接关系,再根据circRNA和miRNA分别在肿瘤中表达差异的方向,保留circRNA与miRNA差异方向相反的链接对,以此构建circRNA-miRNA网络,Cytoscape3.3(http://www.cytoscape.org/)进行可视化(本文所有的网络均采用此软件进行可视化展示)。
②.miRNA-mRNA调控连接关系
miRNA是一类在转录后水平调控基因表达的非编码RNA小分子,成熟的miRNA的长度在22nt左右,通过作用于相应的靶mRNA的3’UTR,下调基因的表达。miRNA与靶mRNA形成了一个复杂的调控网络,在细胞增殖、凋亡、分化、代谢、发育等多种生物学过程中发挥着重要的作用,因此探讨miRNA与靶mRNA的相互作用有助于更好的理解其作用机制、了解基因间的网络调控关系。因此,我们对得到的差异miRNA进行靶基因预测,首先采用TargetScanRelease7.1数据库(http://www.targetscan.org/vert_71/)、MicroCosmVersion5(https://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/)、miRecords(http://mirecords.umn.edu/miRecords)和miRNAMap(http://mirnamap.mbc.nctu.edu.tw/)Version2四个数据库进行预测。保留至少被2个数据库包含的miRNA-mRNA调控关系以增加结果的可靠性。同时,再根据miRNA和mRNA分别在肿瘤中表达差异的方向,保持miRNA与mRNA差异方向相反。此外,再对被miRNA调控的靶标基因,利用BioGRIDVersion3.4.153(http://thebiogrid.org/)、HPRDRelease9(http://www.hprd.org/)和StringVersion10.5(https://string-db.org/),搜索靶标基因之间的相互作用关系,保留至少存在于两个数据集中的互作基因对作为可靠连接。综合miRNA-mRNA调控和靶基因之间的互作关联,构建miRNA调控靶标基因网络,并通过Cytoscape3.3进行网络可视化。最后我们利用DAVID6.8(https://david.ncifcrf.gov/)在线软件,将网络中的基因富集到GO生物学过程和KEGGpathway,识别与靶标基因显著相关的功能和通路,选取p小于0.05作为显著性阈值。
6、预后显著相关的ceRNA网络构建
从TCGA数据库(https://gdc-portal.nci.nih.gov/)中下载某疾病mRNA和miRNA样本,通过样本barcode信息对应后,最终得到配对的mRNA和miRNA具有生存预后信息的肿瘤样本共166个,提取出miRNA调控网络中包含的miRNA和基因在对应样本中的表达值,利用R3.4.1语言中survival包(Version:2.40-1)(https://cran.r-project.org/package=survival)中的单因素cox回归分析筛选预后显著性相关的miRNA和mRNA,再综合circRNA和miRNA之间的连接关系以及miRNA与靶基因间的调控关系,构建与预后显著相关的ceRNA调控网络,并用Cytoscape进行网络的可视化。最后我们利用DAVID6.8在线软件,将ceRNA网络中的基因富集到KEGGpathway,选取p小于0.05作为显著性阈值。
7、某疾病关键因子的识别
首先从miRWalk2.0(http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/index.html)数据库中下载miRNA关联的通路,从中挑选出ceRNA网络中涉及到的miRNA相关的KEGG通路,并与之前一步中得到的ceRNA网络中基因显著富集相关的通路进行比较,取其交叠部分,构建重要通路相关的ceRNA网络,并对网络中的靶标基因进行Kaplan-Meier生存曲线分析。
