3、从返回的结果中找到Output files,下载我们需要的pdb文件
文件①是输入的pdb文件(我们输入了PDB ID,POCASA自动从RCSB PDB库中下载蛋白文件),文件②是我们需要的输出结果,包含了若干潜在口袋的位置信息。将两者下载下来,然后使用PyMOL或其他分子图形软件观察分析。
(POCASA的输出文件,其中XXXX_TopN_pockets.pdb是预测的口袋位置)
另外,在Rank order栏目下,POCASA还告诉我们一共生成了多少个Pocket,每个Pocket都有自己的编号,按照体积排序,依次是Rank 1、2、3……通常,体积最大的Pocket最有可能是真正的蛋白口袋,但体积太大也有可能是假口袋。最保险的做法是进行可视化分析。
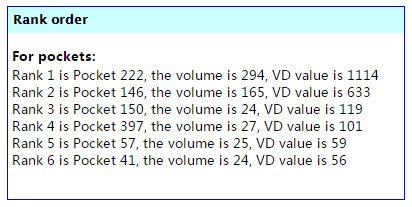
(POCASA计算的口袋体积和口袋可能性排序)
4、使用PyMOL可视化分析
打开1uwh.pdb和1uwh_TopN_pockets.pdb文件,隐藏冗余结构,以cartoon形式显示蛋白A链,以sticks形式显示配体,以spheres形式显示pockets。
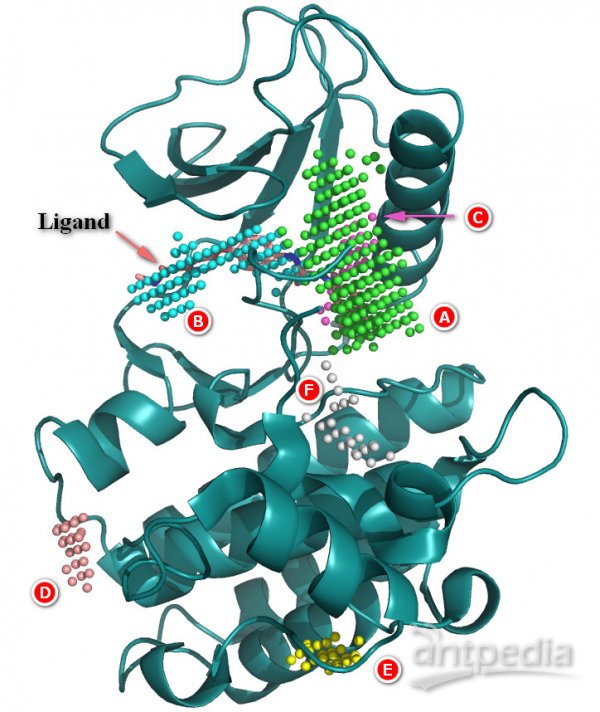
(蛋白是深绿色条带,配体是粉橙色棍棒,Pocket A~F用各种颜色的小球表示)
可见,体积最大的Pocket并不完全是配体的结合口袋;Pocket A只有大约一半体积与配体重叠,Pocket B与配体分子大部分重叠,两个Pocket共同构成配体结合口袋。正如本例所示,我们不能完全相信软件预测的结果,只看体积大小,可能会判断失误。在实践中,应花时间去考察各个预测口袋。
四、 人工观察法
对蛋白口袋/配体结合位点的准确识别,离不开人工观察和分析,仅凭软件预测就匆匆下结论是非常危险的。上面使用POCASA预测1uwh蛋白的口袋位置,Pocket A和B正好组成配体的结合口袋,其实这里面存在一点trick。根据“诱导契合”理论,在配体结合过程中,蛋白与配体都会发生不同程度的构象调整,以达到“最舒服”的状态。这种状态与游离蛋白(free protein,无配体结合的蛋白)是有差异的。上述示例使用了实际上是复合物的蛋白进行预测,成功概率会更大。但实际情况中,需要用到口袋预测的蛋白往往是不含配体的。因此,我们不能期望软件预测的结果总能如示例那样显而易见(Pocket A和B的体积显著大于其他)。在一些口袋不典型、存在多口袋的蛋白中,软件很可能预测不出有效的Pocket,或者预测出多个Pocket。这就需要人工观察去排除可能性极低的Pocket、保留可能性高的Pocket。
续上例,使用PyMOL显示蛋白的(范德华)分子表面。通过观察,我们发现,两个Pocket各自形成亚口袋,配体分子横跨两者。Pocket A开口足够大,并与Pocket F共同形成一个极大的空间区域;Pocket B较为典型,深而且窄;其他Pocket要么体积太小,要么几乎完全暴露在溶液中,不适合充当口袋。因而,Pocket A和B都是有潜在口袋。如果没有配体分子,我们并不能确定哪个才是真正的口袋,或者两者都是或不是。这是普遍存在的情况。严谨的做法是(按照上述标准)排除明显不行的Pocket,把剩下的作为候选口袋,留到后续研究(比如分子对接)中再进一步排除或识别。
还记得上面给出的寻找口袋的原则吗?没错,配体的结合需要疏水作用,通常来说,疏水性空腔更有可能成为口袋。通过蛋白的疏水性分布表面,可以进一步判断口袋的可行性。当然,蛋白内部通常是疏水性的,从形状和位置上也能大致判断某个预测的口袋的疏水性是否足够。PyMOL做疏水性分布表面不怎么方便,我们就忽略这一步骤了。另外,对接打分在一定程度上也能反映口袋的疏水程度,可据此筛选蛋白口袋、识别正确的结合位点。
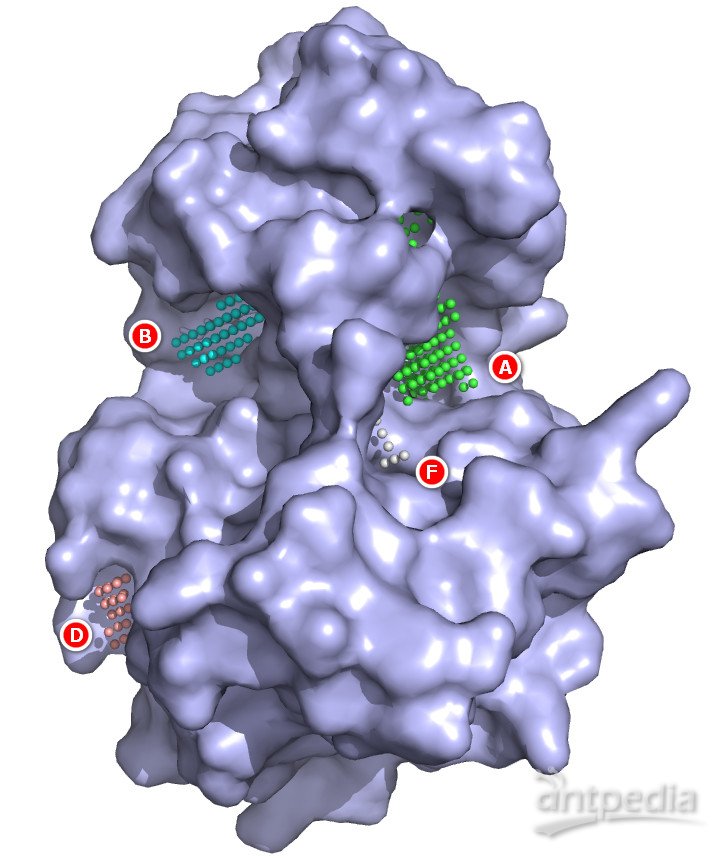
(浅紫色的蛋白表面显示出大大小小的空腔,预测的Pocket小球指示潜在口袋的位置)

 首页
首页






