近日,专注于核酸分子杂交底层技术研发和应用转化的创新型分子诊断公司阅尔基因与同济大学附属上海肺科医院任胜祥教授合作在Nature Communications再次发表重磅研究,介绍了一种多重PCR引物设计的核心算法SADDLE,在大型扩增子法测序panel中首次实现了超低的引物二聚体水平。SADDLE在提高中靶率的同时也降低了测序成本,进一步扩展了相关技术在临床实践中的应用范围。

研究背景
高通量测序(NGS)在遗传疾病、肿瘤驱动基因、宏基因组测序以及鉴定病原微生物等多个领域可以帮助研究者高效地获得基因组信息。当关注的基因数目不是很多或者目标区域占基因组的比例不高时,全基因组测序并不是最优选择,使用靶向捕获或者多重PCR建库将是更好的选择。多重PCR方法操作步骤相对简单,对样本的需求量较低,在生命科学研究和临床应用中均具有广泛应用。此外,多重PCR还可以实现相比连接酶方法更高的测序接头(Adapter)连接效率,针对cfDNA、穿刺样本、胚胎植入前检测(PGT)样本等DNA含量有限的情况尤其适用,相关细节在阅尔基因近日发表的另一篇文章(Ensemble of nucleic acid absolute quantitation modules for copy number variation detection and RNA profiling)中进行过论述。然而,扩增子测序常常难以应用到覆盖上百基因的大型panel当中,其中很重要的原因在于非线性增长的引物二聚体会降低NGS数据的mapping率,进而增加实际的检测成本。针对这一问题,主流的做法有:1.通过在引物中添加经修饰的碱基以便酶解去除;2.根据片段大小进行筛选去除。显然,这类补救措施都有一定的局限性。相反,从源头抑制引物二聚体产生的系统性研究工作却鲜有报道。
众所周知,超多重PCR设计相比单重PCR要复杂的多,更多引物对的加入常常导致体系中的引物二聚体呈爆炸式增长,例如,N重的PCR,引物二聚体可以有C(2N,2)种组合;而M个位点引物则可以有M2N种组合,穷举所有可能性以寻找最优解显然不现实。在数学上,多重引物设计是一个具有高度非凸适应值曲面(non-convex fitness landscape)的高维问题,标准的凸性优化算法(比如gradient descent)并不适用。因此,从底层角度出发,开发一个节约计算资源的、引物二聚体形成少的多重引物设计算法对于将多重PCR应用的靶向高通量测序,进一步减少基因检测所需样本量具有重要意义。
研究内容
该最新项研究开发了一个全新的设计多重引物的算法——二聚体似然估计的模拟退火设计(SADDLE),可以高效地设计多重PCR的引物,有效地降低引物二聚体形成,大大扩展扩增子测序的应用场景。SADDLE的过程包括以下几个步骤:
(1)生成引物池:扩增子测序的PCR产物最短不能短于待测位点的序列,最长不能长于NGS测序模式的读长。3’端位于最短序列之外、5’位于最长序列之内的引物,其扩增产物均可满足扩增子测序的要求。基于该原则,可获得一系列长度不等、与模板的结合能量不等的预用引物池。研究者的前期研究证明当引物和模板结合的ΔG°≈−11.5 kcal/mol时可以很好的平衡效率和特异性。此外,引物太短与模板的结合不稳定而导致扩增效率下降;引物太长则更可能与基因组的其它序列匹配而产生非特异扩增。进一步地,从预用引物池里挑选大小合适,ΔG°介于-10.5~-12.5 kcal/mol,并且GC含量介于-0.25~0.75的引物对,生成引物池。
(2)选取初始引物:针对每一个待扩增序列选取一对初始引物。
(3)计算初始引物的损失函数(Loss function,L(S)):损失函数是计算引物形成引物二聚体的函数,针对选取的引物两两之间的损失值(Badness)总和。其计算公式如下:
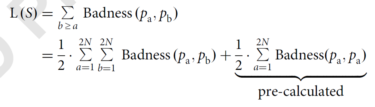
第一项是引物两两之间形成异源引物二聚体的损失值总和,第二项是同一条引物形成同源引物二聚体的损失值总和,该损失值是在设计引物的时候就计算好的。损失值的计算公式如下:
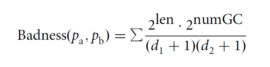
损失值在任意两条引物之间形成4个或4个以上连续的碱基互补配对的序列,容易形成引物二聚体的情况下进行计算。Len碱基互补配对的碱基数,d1和d2是各自引物的互补配对序列与引物的3’的距离(单位:碱基数),numGC是互补配对序列里的GC碱基数。此处选择4作为阈值是因为研究团队在前期研究中表明3个碱基的互补配对并不影响PCR反应。该算法只关注了互补配对序列与3’端的距离,而没有关注与5’端的距离,是因为PCR反应对3’的匹配要求大于对5’端的匹配要求。计算损失值步骤是SADDLE最消耗资源的步骤。
对于损失值的评估,可以如下理解:两条引物之间的互补配对的序列数越多,离3’端越近,GC含量越高,这样的引物对的损失值越高。损失值越高,形成引物二聚体的可能性越大,损失值越低,形成引物二聚体的可能性越小。
(4)生成临时引物:随机选取任意一条引物或一对引物。
(5)计算临时引物的损失函数:用上述公式对临时引物计算损失函数,并与上一次选取的引物的损失函数进行比较,如果临时引物的损失函数更小,那么这条/对临时引物就会迭代上一条/对选取的引物。
(6)重复步骤4,5,直至满足预设的边界条件。研究团队一般推荐多设置几个初始引物,以确保获得最佳的引物组合。
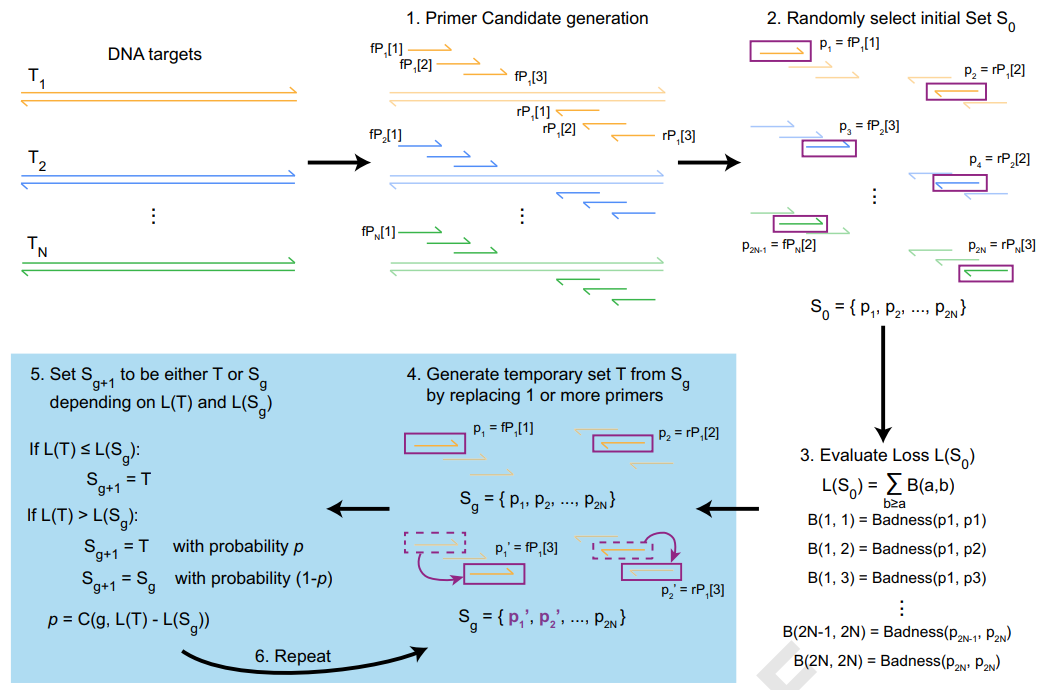
图1.SADDLE算法的基本流程
接下来,研究团队尝试将SADDLE应用到一个靶向肿瘤相关基因的96-plex panel中。整个过程在一台普通PC仪上仅耗时10分钟左右。作为对比,团队一共合成了未优化(PS1)、部分优化(PS2)、饱和优化(PS3)三套引物组合进行实验验证。

图2.基于SADDLE框架的多重引物设计
首先以gDNA作为样本进行实验,文库电泳结果显示,优化程度越高,目标条带越清晰。最终的测序数据也可以看出,引物二聚体的比例从PS1的90.7%下降到PS3的4.9%。随后,在多个FFPE临床样本中也获得了类似的结果。研究团队进一步设计了384-plex panel(768 引物)进行验证,优化流程耗时60分钟,而最终实验观察到的引物二聚体比例仅为1%!
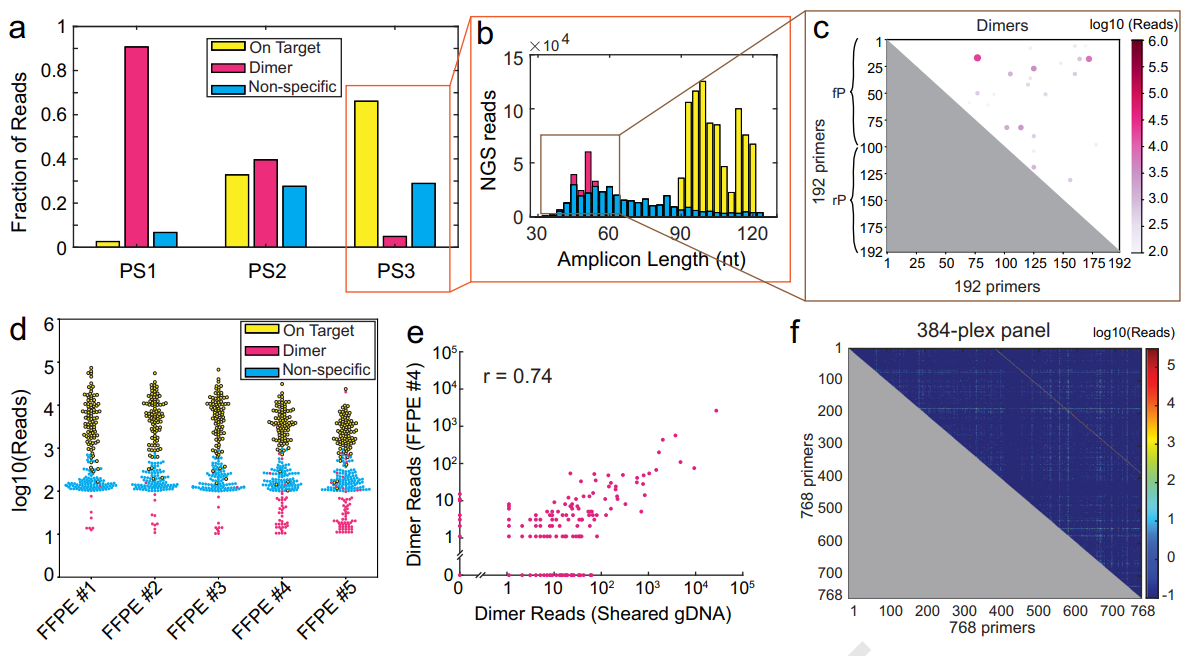
图3.基于SADDLE设计的NGS引物组的验证结果
通常,研究人员需要在算法精确度和计算复杂度之间做出取舍,因此,准确评估损失函数的预测性能对于进一步优化多重PCR引物设计至关重要。通过前面的实践,可以总结出函数预测值与实际观测的二聚体reads数之间的关系。当处于最佳平衡点时,灵敏度和特异性分别为92.5%和90.3%。通过改变函数阈值,得到的曲线下面积(AUROC)为0.9577,显示出SADDLE良好的预测能力。需要注意的是,实验过程中观察到一些很强的二聚体没有被准确预测,说明即使忽略计算复杂度的限制,仅靠算法依然很难完全消除全部二聚体。
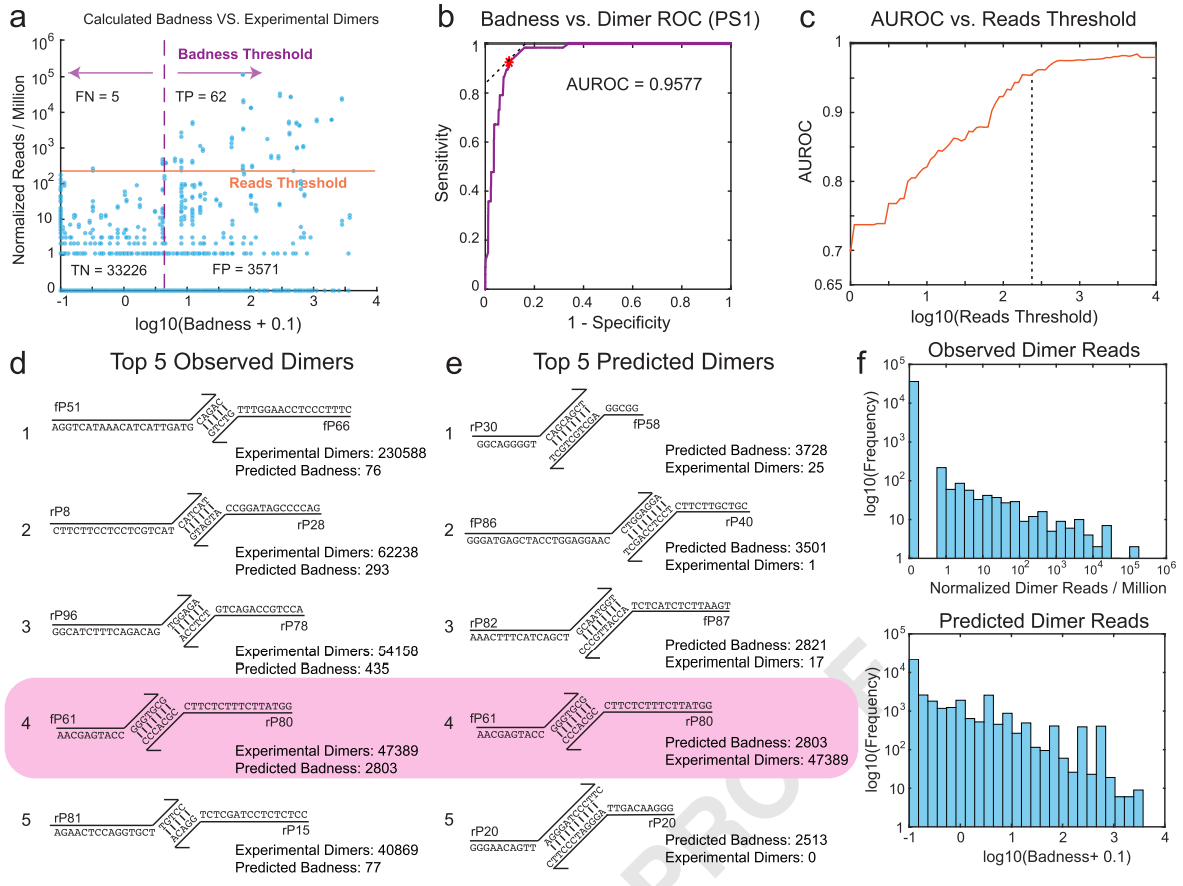
图4.损失函数对单个引物二聚体的预测准确性分析
最后,研究团队测试了SADDLE在基因融合检测技术中的表现。基因融合是靶向治疗的重要靶点,因此在用药指导中是重要的诊断标志物。通常通过单通道的qPCR对已知融合位点进行检测或用NGS进行检测。针对多个位点一次性的进行多重qPCR可以大大提高基因融合检测的可操作性。该研究团队针对非小细胞肺癌(NSCLC)6个常见的融合基因的56个融合类型设计了多重qPCR引物组。以非小细胞肺癌病人的RNA反转录成的cDNA作为样本,证明了该引物组可显著区分融合基因与野生型,即使在低至1% VAF的模拟样本中也可以成功检测。另外,从NSCLC患者的血浆外泌体中提取的RNA中也成功检测到相关融合事件。
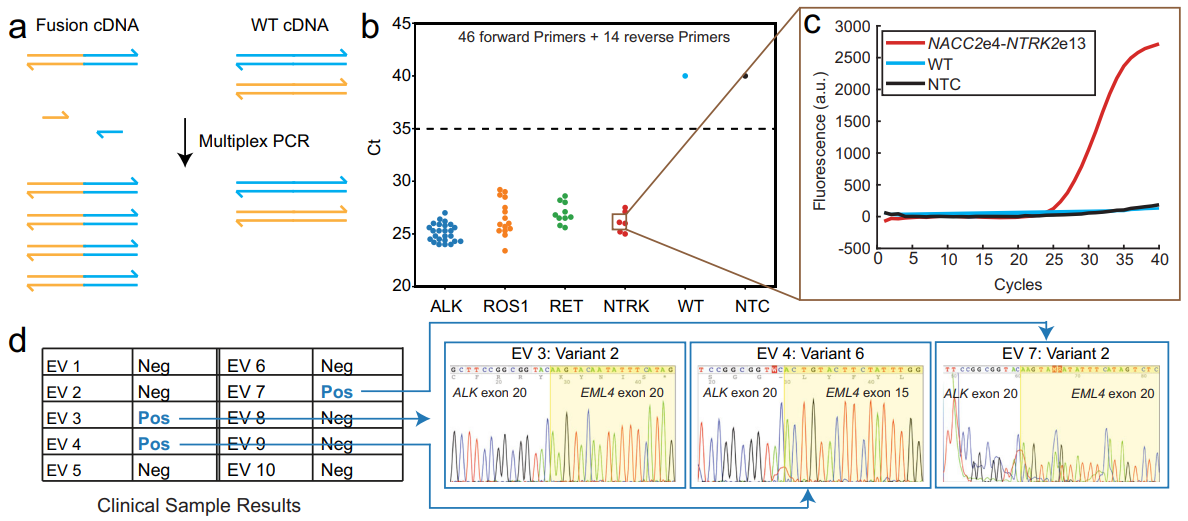
图5.基于SADDLE设计的qPCR引物组的基因融合检测
结 语
总之,研究团队提出并验证了一种用于优化多重PCR引物的算法框架——二聚体似然估计的模拟退火设计(SADDLE),相比常规流程大大降低了引物二聚体的形成。在一个96-plex panel中,SADDLE的引入将产物中二聚体的比例从90.7%降低到4.9%,中靶率及扩增子的均一性也得到提升,即使扩展到384-plex依然能够将这个比例维持在较低水平。因此,SADDLE无需额外的二聚体清除步骤即可提高reads利用率,进而节约大panel的试剂成本和测序成本。扩增子测序更节约样本,实验周期更短,因此针对样本珍贵并且时效要求高的应用场景,例如肿瘤病人的穿刺样本、血液的cfDNA,羊膜穿刺样本,胚胎植入前筛选(PGS)和胚胎植入前诊断(PGD)等,扩增子测序有显著的优势。降低引物二聚体的形成可以大大提高扩增子测序的检测位点,大大提高扩增子测序的适用场景。
除了NGS之外,该团队还将SADDLE方法应用到qPCR当中,实现了低成本的融合基因超多重检测。与此同时,非特异性扩增的问题在低引物二聚体的背景下凸显出来,该团队将据此进一步优化算法,挑战更大型的扩增子测序panel。
原文链接:https://doi.org/10.1038/s41467-022-29500-4
作者点评
“NGS panel中引物二聚体的形成会降低mapping率,既增加成本也降低了测序灵敏度,”文章第一作者Nina G. Xie表示。“通过算法设计出低引物二聚体的超多重PCR引物组,SADDLE解决了扩增子测序文库制备方法的关键瓶颈之一,并且简化了靶向测序panel的实验流程。”靶向测序或者qPCR方法都被广泛用于检测疾病相关的DNA突变,前者能提供更丰富的信息,但成本高、周期长,qPCR相比而言更简便快速,但能提供的DNA标志物信息却很有限(通常小于6个)。SADDLE的出现可以大大提升这两个平台的能力。
“借助SADDLE,我们设计并验证了一个引物二聚体比例显著下降的靶向测序panel,” 该研究的共同通讯作者、同济大学附属上海肺科医院任胜祥教授提到。“此外,我们也设计了单管的多重qPCR方案,可以同时检测56个不同的基因融合,并且通过了临床样本验证。”
“我们认为qPCR和靶向测序panel对于精准医疗是同等重要的工具,” 该研究共同通讯作者、阅尔基因美国创新中心负责人David Zhang表示。“前者提供了一种经济快速的检测手段,后者可以给出全面的基因组信息进而辅助治疗。阅尔基因致力于相关技术的开发与转化以推动分子诊断方法的发展,造福人类健康。”
关于阅尔基因
阅尔基因长期专注于核酸分子杂交底,层技术原理的研究,在分子检测领域发表了众多硬核研究成果。早在2012年,阅尔基因团队即提出了一个分析核酸杂交特异性的理论框架,相关成果发表在权威期刊Nature Chemistry上。2015年同刊发表的新研究构建了一个基于杂交过程的动力学反应模型来预测最佳热力学参数,同时证明这种动力学模拟对实现高特异性是必要的。2017年再次报告了一种加权邻近表决(weighted neighbour voting, WNV)预测算法,可以基于序列信息精确计算核酸杂交反应的速率常数,助力复杂多重体系的DNA探针设计。去年,团队克服了多重PCR的诸多难点,在Nature Biomedical Engineering公布了独创的mBDA技术,将超低频变异检测扩展到多重panel。


 首页
首页






