
原文链接:https://doi.org/10.3390/molecules28041557
摘要:
通过UHPLC-HRMS对Cortinarius ominosus担子果的提取物分析发现,该蘑菇中含有一种未见报道的特殊代谢产物,其分子式为C17H10O8,其H/C原子比例极低,使得该化合物的结构鉴定工作非常具有挑战性。这种天然产物的化学结构——即Ominoxanthone(1),一般不能仅依靠传统的1D/2D NMR数据来解决,因为这些数据提供的信息非常有限,无法推导出明确的结果。然而,我们采用了计算机辅助结构解析(Computer-Assisted Structure Elucidation,CASE)工作流程,对所有与其NMR数据一致的结构进行打分排列,以解决这个问题。在一组排名靠前的结构中,基于密度泛函理论(Density Functional Theory,DFT)的化学位移计算进一步确定了化合物Ominoxanthone的化学结构。虽然Ominoxanthone的分子骨架在天然产物中是前所未有的,但我们提出其合理的生物合成途径,其中涉及cortinariaceous源的前体和经典的生物合成反应。
1、简介
在先前有关真菌代谢产物的研究工作中[1],我们通过UHPLC-HRMS对Cortinarius ominosus的DCM/MeOH (1/1, v/v)提取物进行了分析,发现这种蘑菇富含有一种物质,在m/z 343.0450处检测到明显的信号,元素组成为C17H10O8。值得注意的是,我们在《Dictionary of Natural Products》数据库[2]中检索该分子式,没有任何匹配结果,这促使我们对其进行进一步的结构阐明研究。尽管这种精确的分子式以前尚未被报道过,但它让人联想到蒽醌类色素,这类化合物经常被报道来自于cortinariaceous 种属[3]。据了解,Cortinarius属以及Dermocybe亚属的蘑菇可以产生大量的羟基蒽醌、蒽醌前体和吡喃萘醌类代谢物,从而使它们的子实体呈现黄色、橙色或红色[4]。本次所研究的化合物,即Ominoxanthone (1),可以通过制备性高效液相色谱从原始提取物中简单地纯化出来。
2、结果与讨论
针对化合物1 的研究,我们首先碰到的一个困难是——它在大多数常见的 NMR 氘代溶剂中溶解度很低。这让我们感到意外,因为从 Cortinarius spp. 提取出的众多聚酮类色素都可以在常见的氘代溶剂中测试其 NMR 谱。我们通过筛选不同的溶剂,最终发现氘代三氟乙酸(TFA-d) 是能促进化合物1 溶解和获取 NMR 数据的最佳选择。另外,DMSO-d6 也提供了较好的 1H 和 13C NMR 数据,但 1H NMR 信号过宽,得不到令人满意的 HMBC 谱。
化合物1 的1H NMR谱图显示存在三个芳香质子——δH 7.47(1H,s),7.53和8.00,后两者为间位排列(1H both,d,J = 2.3 Hz),一个连氧亚甲基(δH 5.82(2H,s)),以及一个甲氧基(δH 4.34(3H,s))。通过13C NMR谱与HSQC谱相结合分析,推测存在三个羰基,三个芳香甲基,一个连氧亚甲基,一个甲氧基,以及九个非质子化的芳香碳原子(包括四个连氧碳原子)。结合其分子式分析,这些NMR数据提示,化合物1 应该是一个甲氧基化的八元酮类化合物,与Cortinarius种属中大多数聚酮类物质的结构一致。从生物合成的角度看,经典的八酮基类蒽醌是由一个乙酸起始物和七个丙酸盐单元的线性前体组装而成的。线性多酮前体的区域选择性真菌环化模式,即所谓的F模式,将产生两个奇数侧链,从而得到典型的内黄素骨架(图1A),尽管C-2处的频繁脱羧通常会导致最终代谢物只显示一个奇数侧链[6]。因此,在开始对1的结构鉴定前,我们假设连氧亚甲基应连接于A环上的C-3位置(图1)。

图1.(A)内黄素的化学结构,真菌聚酮类化合物的生物合成基础。(B)蒽醌类色素noraustrocorticin的化学结构,来自未确定的澳大利亚Cortinarius物种。(C)1A—在含氧杂环二酮的苯醌二甲酸酐骨架上的关键2D NMR相关性。(D)1B—在黄酮骨架上的关键2D NMR相关性。
取代基的相对位置可以从ROE实验中推断,将甲氧基放置在两个间位的芳香族质子之间,后两者之间的COSY信号进一步证实了这一点。由于这个自旋系统不涉及亚甲氧基信号,因此推测这些信号与C环有关(图1A)。这些谱图信号特征确实让人联想起许多聚酮色素结构的C环结构,它们通常包含这种取代模式,占据C-5、C-6和C-7位置(图1A),但不确定是C-5还是C-7的位置[5,7,8]。考虑到分子式要求和化合物1 的聚酮来源,我们推断一个-OH基团应该位于C-8,即使这个假设没有谱图数据支持。尽管一般认为在cortinariaceous聚酮类化合物中,氧取代基都在C-8位置,但将其C环核磁共振(NMR)信号与C环同样带有取代基的蒽醌的信号进行比较,发现NMR光谱数据的相似性非常有限。特别是,与先前的蒽醌类化合物相比,化合物1 中芳香环中次甲基的13C NMR化学位移的差异非常明显,前者这两个位置的信号几乎相等[5,7]。对于另一个暂定位于A环上的自旋系统,在氧化的亚甲基质子H2‐3′和第三个孤立的芳香质子H‐4之间可以观察到ROESY和COSY信号。HMBC核磁信号表明,氧化的亚甲基质子H2-3''与羰基碳原子δC 176.3(2-COO),和两个芳香季碳原子(δC 108.6(C-2)和158.8(C-3))以及δC 104.0(C-4)的芳香甲基形成一个苯环并γ-内酯结构片段。这些核磁共振信号特征指向类似天然产物 Noraustrocorticin(图1B)的结构[5]。这些亚结构核磁共振数据与Noraustrocorticin得到的数据之间具有良好的一致性,进一步支持了这种推测,也表明应该将一个-OH基团定位在C-1,与这个环必须五取代和分子式要求相符。从生物合成的角度来看,这种假设是合理的,但缺乏直接的光谱证据。即使在没有直接光谱支持的情况下引入了两个酚羟基,仍然需要在结构1上放置三个氧原子以满足分子式要求。这排除了化合物1具有蒽醌骨架的可能性。有趣的是,对HMBC光谱的粗略检查显示,在δH 7.53 (H‐5或H‐7)和δH 7.47 (H‐4)处的芳香质子与δC 184.1(C‐9)的羰基碳原子之间存在4J 的相关,可以推断化合物1包含一个苯甲酮骨架。最直接完成该结构的拼装策略是,根据得到的分子式,通过连接两个苯环形成一个内酯环,将C-4a和C-10a联系起来。这种连接将导致一个氧杂环戊二烯二酮型B环,提供第一个结构假设——1A(图1C)。这种暂定的骨架结构在谱图特征上与蒽醌不一致,结果是令人满意的。因此,从芳香族质子H‐4到δC 163.4 (C‐4a)的氧合sp²碳的HMBC相关性,与毗邻的氧原子的引入相一致,这也得到了从H2-3''到同一碳的4J HMBC相关性的支持。同样,从C环信号观察到的长程杂核相关性与1A结构一致。在δH 8.00处的第一个芳香性质子(在此假设下为H-5),与C-6和δC 175.2处的羰基碳原子存在HMBC相关,似乎可以归属于内酯环的C-10,而第二个芳香性质子在δH 7.53处(暂定为H-7)显示与C-6以及δC 162.30处含氧sp²碳原子存在HMBC相关(与先前提及的与C-9的4J相关性一致)。尽管这些数据完全适用于结构1A,但我们可以构想一个符合所有上述光谱特征的替代结构1B(图1D)。该结构包括一个黄酮骨架和一个在C-8位置的羧酸基。同样,从在δH 7.53处检测到的芳香性质子(在这里对应H-5)到在δC 162.3处检测到的含氧sp²碳碳原子之间的HMBC相关将代表与C-10a的相关性。同样,从在δH 8.00处的第二个芳香性质子与δC 175.2处碳原子的HMBC相关可以通过H-5到8-COOH的相关性来解释。
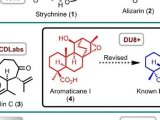
一项文献调查证实,区分这两种骨架相当困难,因为这两对化合物显示相同的碳连接,因此HMBC相关性数据相似[9]。有趣的是,含有这种氧杂环己二酮(oxepinedione)的代谢物据称曾多次从真菌来源中分离出来,主要来自微真菌(micromycetes)[10-12]。事实证明,许多这样的代谢物最初被错误地阐明,后来被修改为含羧酸的呫吨酮类化合物(xanthones)[9,13]。举例说明,化合物wentiquinone C的结构修正概述如下(如图2)。
图2.化合物wentiquinone C的结构修正
在整个文献中,呫吨酮类化合物和二蒽醌类化合物(seco‐anthraquinones)的NMR数据的微小差异至少被强调了两次,导致了批量结构需要进行修正[9,13]。这些修正依赖于化学衍生化的策略,基于对甲基化衍生物的NMR标志的粗略分析。因此,甲基化后甲氧基酯相关共振信号(δC< 55 ppm)的出现可能表明了游离羧酸的存在,因此可能不参与环化过程,从而证明是一个呫吨酮骨架分子。在更简单的情况下,天然产物中本来就存在一个甲氧基酯基团,但它被错误地理解为芳香性甲氧基,有时会导致将其结构错误地解析为氧戊二酮衍生物,而不是正确的含有甲氧基酯‐呫吨酮类分子[9,13]。
有趣的是,1B与1A的结构差异类似于氧戊二酮与其修正后的呫吨酮结构(图2)。1B比1A的结构更合理一个证据是,化合物1 的NMR波谱数据与羧酸呫吨酮的NMR波谱数据之间更吻合(图3)[9,14]。

图3.1B的C环相关的13C NMR化学位移与wentiquinone C的化学位移进行比较(用于比较的化学位移来自DMSO‐d6获得的NMR谱)。
总的来说,对化合物1的核磁共振波谱数据粗略检查,会引导我们将Ominoxanthone的结构解析为1B的合理结果。然而,从这个分离物获得的关键核磁共振实验的信息相当有限,因为它的质子缺乏的性质,使结构的阐明变得曲折。通过所谓的Crews规则可知,当H/C原子的比率小于1时,基于NMR的结构阐明有很高的不准确的风险,就像这里的情况[15]。从这些结构的NMR数据中获得的信息很少,很可能会导致出现几个可能的候选结构,因此确保已经充分考虑了所有可能的结构非常重要 [16]。
为了验证我们的结构假设,我们选择通过ACD/Structure Elucidator软件[18],将化合物1 的核磁共振波谱数据提交给计算机辅助结构解析(CASE)工作流程[17]。简而言之,该程序利用高分辨质谱推导的分子式和1D (1H和13C)和2D (COSY, HSQC和HMBC) NMR谱图数据来创建分子连接图(MCD)。MCD包含了所有原子个数以及它们的化学位移、性质(价态、杂化状态和相邻杂原子的可接受性)等信息。COSY相关信息——诊断一个C-C键的长度(3JHH),和HMBC相关性——推测指示一个或两个C-C键的距离(2,3JCH),这些信息也一并被记录在MCD中。基于MCD中包含的数据,软件可以生成满足所有满足指定分子式和1D和2D NMR数据的结构异构体。随后,软件基于谱图知识和结构过滤系统对暂定的结构候选库进行过滤[17]。在一组生成的候选结构中,软件基于三种经验算法(增量法、神经网络算法和HOSE算法)对其13C NMR化学位移进行预测[19,20],然后选择最可能的结构,因此,通常最合理的结构预测误差在1.6 ~ 1.8 ppm范围[17]。根据这一步的化学位移预测,CASE系统按照化合物实验和预测的NMR化学位移之间的差异递增排序(通常表示为平均偏差),输出的候选结构。如果这种基于经验预测算法没法成功地明确区分正确的结构,那么可以通过更高精度的量子力学方法对这些排名靠前的结构进行NMR计算[21,22]。
在溶剂TFA-d中获取的化合物1的光谱数据被ACD/SE程序用来生成一个MCD(如图4),通过该MCD严格的结构生成工作流程,可以在31分钟内提出八种可能的结构(如图S14)。

图4. 从在TFA-d 溶剂中记录的ominxanthone(C17H10O8)的NMR数据中获得的分子连接图。
sp3和sp²杂化的碳原子分别用蓝色和紫色显示。连接杂原子的可接受性由标签“ob”(强制)和“fb”(禁止)表示。HMBC相关性由绿色箭头表示。紫色箭头指的是非标准相关性(nJHH,CH长度n>3,)。这些相关性被设置为弱HMBC信号。

图S14. 根据碳谱化学位移偏差递增顺序排列的8个候选结构
这些结构按平均偏差dA、dN和dI的递增顺序排列,如图S14所示。自动归属化学位移的三个排名最高的结构如图5所示。图5中还显示了每个结构的最大13C NMR化学位移偏差。令人高兴的是,排名第一的结构与结构1B相同。有趣的是,备选结构1A竟然是CASE工作流程的第三个候选结构(图5)

图5:三个排名最高的候选结构(1、2和3),按照光谱差异递增的顺序列出。根据预测方法,平均偏差分别报告为dA,HOSE算法;dN,神经网络算法;和dI,增量法,以及每种预测方法的最大13C NMR化学位移偏差。
很不幸,它们实验和预测的13C NMR化学位移之间的平均误差都是在一个水平,并且比通常观察到的这类分析的差异要大(即,dn < 3 ppm,其中n = A、I和N)。由于ACD/Predictors 是基于含有传统溶剂记录的NMR数据库进行训练的[18],因此,该案例中的NMR数据是在TFA-d 中获得的,可能会导致较大的差异。这是一种很少选择的NMR溶剂,但是已知对13C NMR化学位移产生重大影响[23]。我们想评估是否通过将模拟光谱与在DMSO-d6中获取的NMR数据进行比较,可以获得更好的结果,即使由于1H信号变宽,从这些光谱中获得的信息程度相对较低。为了弥补在该溶剂中获得的相关信号较低的水平,我们将从TFA-d 获得的MCD适应于在DMSO-d6中观察到的信号。为此,我们在两个NMR数据集中按化学位移值递减排序并相应地进行转置,除非在DMSO-d6中获得的光谱特征明显相反。虽然这种策略不能保证所有信号归属的正确性,但我们假设溶剂变化时交换的信号应该显示相似的化学位移值,因此该策略应该产生适度的化学位移误差。从在DMSO-d6中获得的NMR数据创建的MCD如图S15所示。通过ACD/SE软件进行结构生成,获得了20种结构,用时34分钟。前三个排名最高的结构如图6所示。

图6. 三个排名最高的候选结构(1、2和3),其13C化学位移在DMSO-d6中测量。偏差的标识与图5相同。颜色编码如下,表示13C NMR化学位移预测的准确性:绿色=理论值与实验值之间的差异<3 ppm,黄色=差异介于3和15 ppm之间,红色=差异超过15 ppm。
结果再次证实了1B是首选的结构(见图6)。令人满意的是,这使用更普遍的NMR溶剂所得到的偏差大小处于正确结构所获得的典型范围内。值得强调的是,第二个首选的结构,即"角形"呫吨酮结构,与使用TFA-d作为溶剂所获得的NMR数据执行CASE工作流程时的结果相同。我们对这个结构持怀疑态度,因为这样的"角形"呫吨酮结构并不符合从Dermocybe/Cortinarius蘑菇报道的蒽醌羧酸中不断发现的内环蒽醌骨架[3,24]。通过DP4概率的计算,结果表明结构#1(图6)是最合理的:DP4A(13C)= 95.29%,DP4N(13C)= 99.5%,DP4I(13C)= 100%。
尽管如此,在这种情况下,通过DFT方法对该候选结构进行13C化学位移的预测,仍然值得推荐的策略方法[21,22]。我们对CASE系统推荐的3个最优结构进行准确的DFT-NMR计算,以进一步判断区分(表1和表2)。这些QM计算的总结在补充材料的图S16中提供。基于13C NMR化学位移的最小均方根偏差(RMSD)和最低最大偏差(Max_dev)计算结果,DFT-NMR预测结果表明,结构#1(即1B结构)为最合理的结构。
表1:化合物1的1H 和13C NMR (700/175 MHz) 数据
a注意,这种溶剂下获得的NMR数据未通过2D NMR数据验证,因此提供的一些赋值可能会颠倒。
表2:以下是在Figure 5和Figure 6中展示的ominoxanthone三个排名最高的候选结构的实验和DFT计算的13C NMR化学位移(ppm)(为了清晰起见,所提供的编号适用于排名最高的候选结构;其他两个候选结构的每个碳的化学位移归属信息在Figure S16中给出,附录材料):

本次报道的天然产物Ominoxanthone 是首次发现与γ-内酯结构线性融合的呫吨酮类化合物(xanthones)。该化合物与以前报道的从cortinariaceous种属[5]中分离的蒽醌类化合物相似。从蒽醌到呫吨酮的生物合成相互转化具有相当的泛化性[13,14,25],因此,去甲基化的noraustrocorticin衍生物的去甲基化被认为是Ominoxanthone的生物合成途径(图7)。有趣的是,Steglich等人之前报道了从Cortinarius cotoneus分离得到化合物Leprocybin [26],该化合物是线性融合吡喃酮环的呫吨酮结构。Leprocybin还与一种蒽醌类化合物dermolactone在结构上有重要的相似性,后者在几年后被Gill和Gimenez报道[7]。
图7. Ominoxanthone的合理生物合成途径
3、材料与方法
略
4、总结
在此,我们通过CASE和DFT计算策略阐明和验证了一个新型呫吨酮类化合物——Ominoxanthone。该化合物的结构与从Cortinarius sp.中分离的蒽醌类化合物noraustrocorticin和austrocorticin的结构很相似,这是第一个与γ-内酯结构线性融合的蒽醌类化合物。














 首页
首页




