在过去的十年中,RNA测序(RNA-seq)已经成为在全转录组范围内分析差异基因表达和mRNAs差异剪接的重要工具。然而,随着下一代测序技术的发展,RNA-seq技术也在不断发展。现在,RNA-seq用于研究RNA生物学的许多方面,其中包括单细胞基因表达、翻译(翻译组,translatome)和RNA结构(结构组,structurome)。RNA-seq的其它应用也在开发中,例如空间转录学(spatialomics)。加上新的长读长 (long-read,注:在本文中,RNA-seq测序生成的read统一译为“读长“)和直接RNA-seq(direct RNA-seq)技术以及用于数据分析的更好的计算工具的整合,RNA-seq技术的创新有助于人们更全面地理解RNA生物学,例如从何时何地转录发生到控制RNA功能的折叠和分子间相互作用等问题。
RNA-seq技术出现于十年之前,自其诞生之日起,RNA-seq就成了研究分子生物学的普遍工具,这项技术几乎构成了我们对基因组功能的认知基础 。RNA-seq中最常用的分析方法就是找出差异基因表达(Differential gene expression, DGE)。从最早的出版期刊开始,DGE分析的基本阶段就未发生实质性的改变。
在实验室中,其标准流程就分为三步:
第一步是构建测序文库,这一步骤包括提取RNA,富集mRNA或清除核糖体RNA,合成 cDNA,加上接头。
第二步,在高通量平台(通常是Illumina平台)上对文库进行测序,每个样本的测序深度为10-30M读长数(读长这里就是前面说的reads)。
第三步是数据分析,具体的工作是:对测序得到的读长进行比对(aligning)和/或组装到转录组上,对这些覆盖了转录组的读长进行过滤,归一化(Normalization),根据统计模型找出那些在不同样本之间有差异的转录本。早期的RNA-seq从大量的实验样本中产生了DGE数据,这充分说明了RNA-seq在广泛的生物体以及系统中的使用,这些生物体包括玉米(Zea mays), 拟南芥(Arabiodopsis thaliana), 酿酒酵母(Saccharomyces cerevisae),小鼠(Mus musculus)以及人类。虽然RNA-seq这个术语经常被用于那些完全不同的方法学方法和/或生物学,但是DGE分析仍然是RNA-seq(补充材料中的表1)的主要应用,并被视为常规研究工具。
RNA-seq的更广泛应用已经促进了我们对生物学多方面的理解 ,例如通过提示mRNA剪接和非编码RNAs和增强子RNAs对基因表达的调控。RNA-seq的应用和进步是由技术发展(湿实验室和计算生物学)驱动的,相对于以前的基因芯片,RNA-seq这种方法对RNA生物学和转录组产生更丰富并且偏见更小的信息。到目前为止,从标准的RNA-seq方法衍生而来的各种RNA-seq方法几乎有100种。Illumina的短读长(short-read)测序平台能对这些由大部分不同方法的RNA-seq构建的文库进行测序,但是最近长读长(long-read)RNA-seq的与直接RNA-seq测序(direct RNA sequencing, dRNA-seq)的进步已经能够解决以前研究人员使用短序列手段无法解决的一些问题。
在这篇综述中,我们首先会介绍一些最基本的短读长RNA-seq中的DGE方法,再将这种基础方法与最近新兴的长读长RNA-seq和dRNA-seq进行比较。我们会介绍短读长测序方法在文库制备方面的进展,以及实验设计和DGE的数据分析方法。随后我们会拓展这些常规的RNA-seq方法,介绍一些单细胞测序和空间转录组学的分析。我们会提供一些案例,介绍RNA-seq在RNA生物学方面的关键应用,包括转录组分析,翻译动力学,RNA结构,RNA-RNA之间相互作用和RNA-蛋白质的相互作用。最后,我们会简单描述一下RNA-seq的未来,以及单细胞和空间RNA-seq方法是否会像DGE分析一样成为常规工具,长读长测序方法是否会取代短读长测序方法。由于篇幅限制,我们无法介绍所有的RNA-seq方法,在这些方法中,值得注意的是非编码转录组学,原核转录组学(prokaryotic transcriptomes)和表观转录组学(epitranscriptome)。
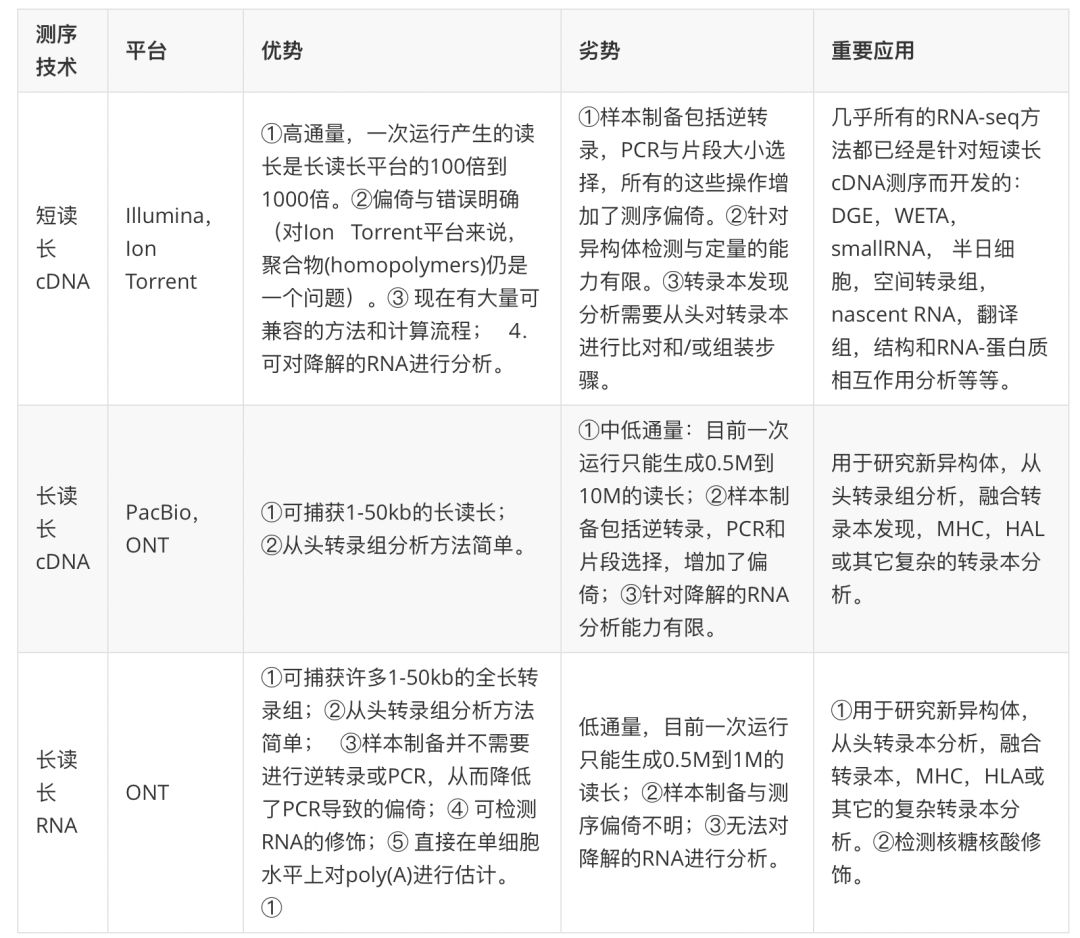
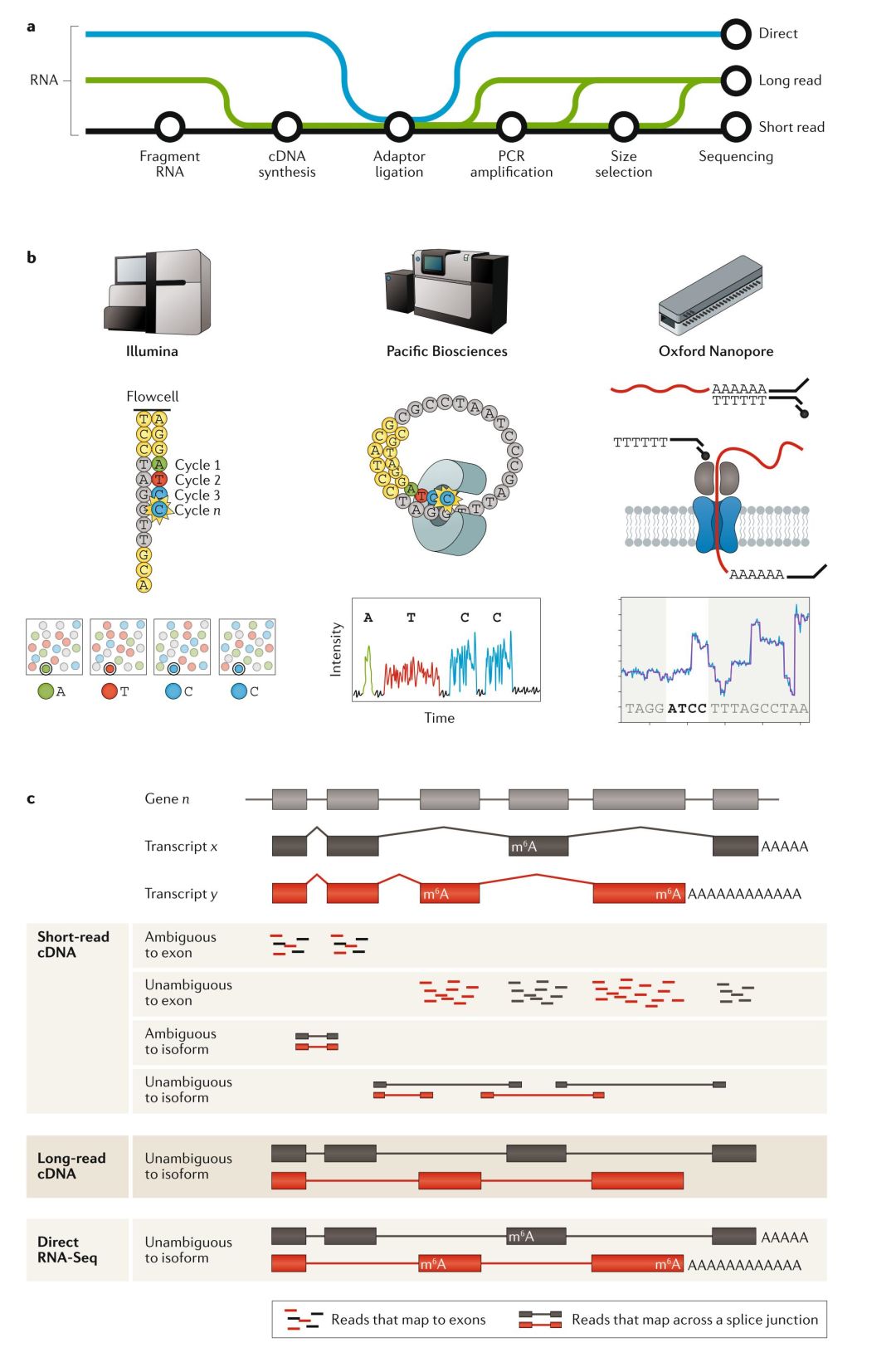
Illumina的短序列读长测序技术生成了SRA(Short Read Archive)中95%已表达的数据(附件表2)。由于cDNA的短序列读长测序方法几乎是一种常规的方法,因此 我们认为这是一种最基础的 RNA-seq技术,我们先来讨论这种测序主要流程与局限。不过,长读长cDNA测序与dRNA-seq已经兴起,随着研究人员对能提供更丰富转录本水平方面(isoform-level)数据需求增大,这两种新的测序方法有望对常规的短读长测序方法提出挑战(FIG1, TABLE1)。





 首页
首页


