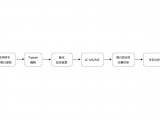
短读长已经成了在整个转录组范围内对基因进行检测和定量的事实方法(de facto method),部分原因是这种方法比芯片成本更低,操作更方便,但是其主要原因还是因为这种方法能生成更全面,更高质量的数据,这种方法能够 对整个转录组中的基因表达水平进行定量。使用Illumina短读长测序平台进行DGE分析的核心步骤包括:RNA提取、cDNA合成、接头连接、PCR扩增、测序和数据分析(FIG1)。在这个过程中,存在打断片段,片段长度选择和基于磁珠的文库纯化这些操作,因此这种方法产生的cDNA片段通常都是在200bp以下。RNA-seq文库的测序读长分配到每个样本上的话,每个样本会测到平均20-30 million条读长(reads)(也就是常说的20-30M条读长),数据经过处理后,使用这些读长对每个基因或转录本进行定量,最后再用统计学方法来统计基因的差异。短读长RNA-seq方法很稳健,并且通过对短读长测序技术的大范围比较发现,这种技术在平台内和平台间的相关性很好。但是,在样本制备和数据分析这两个阶段会引入一些干扰和偏倚。这种局限可能会影响通过实验来解决特定生物学问题的能力,例如准确识别和量化多个异构体中的哪个来源于一个基因。对于研究那些非常长,高度可变的转录本异构的人来说,这种局限表现得尤为明显,例如在人类转录组研究中;人类转录本的长度范围是109bp到186kb,其中50%转录本长度大于2500bp。尽管短读长RNA-seq可以对最长的转录本进行详细的分析,但是涉及的实验方法不能扩展到全转录组分析。其他的偏倚与局限来源于那些大量的计算方法,这些方法包括例如如何处理模糊或多个回贴的读长(multi-mapped reads)。现在出现了一种合成长读长(synthetic long reads)的新方法,这种方法可以实现全长的mRNA测序,并试图解决其中的一些局限。这种方法使用了唯一分子标识符(unique molecular identifiers,UMI)来标记全长的cDNA,在制备短读长RNA文库之前,加入的UMI会随着单个cDNA分子而进行复制。转录本异构体可以在高达4kd的contigs中重建,用于发现异构体和表达分析。但是,对于从根本上解决短读长cDNA测序固有局限的最可能解决方案则是长读长cDNA测序和dRNA-seq测序 。
虽然Illumina测序目前是占主导地位的RNA-seq平台,但PacBio和Oxford Nanopore(ONT)公司都提供了可供选择的长读长技术,能够对完整的单个RNA分子进行单分子水平级的测序。通过消除短RNA-seq测序数据的组装这一步,这些新方法克服了短读长测序方法相关的一些问题。例如,减少了测序读长回贴过程中的歧义,并且可以识别更长的转录本,这样就能获取更完整的异构体多样性信息。这些方法还能降低许多短读长RNA-seq计算工具中关于剪接连接的假阳性。
PacBio的Iso-Seq技术可以读取最高可达15kb的转录本的全长cDNA,这就有利于发现大量以前未注释的转录本,并通过检测物种的全长同源序列证实了早期的基因预测。在标准的Iso-Seq操作流程中,高质量的RNA被一个模板切换凝聚力转录酶(a template-switching reverse transcriptase)反转录为全长的cDNA。生成的cDNAs再经过PCR扩增,加入到PacBio的单分子实时(single-molecule, real-time)文库制备系统中。制备好的短转录本序列可以很快地扩散到测序芯片的活性表面,但由于短转录本的测序存在偏倚,因此在对转录本进行测序时,建议选择片段的长度是1到4kb,这样就能在此范围对长转录本和短转录本进行更加均匀地采样。由于PacBio测序方法需要大量的模板,因此需要进行多轮PCR,不过这一操作还需要进行优化,从而降低扩增导至的偏倚。经过PCR的末端修复和PacBio SMRT接头连接后,就可以进行长读长测序了;通过修改测序芯片的上样条件,就可以在这一步骤进一步控制测序片段长度。
ONT cDNA测序方法也能产生全长的转录本读长,甚至还能在单细胞水平上产生该读长。模板转录逆转录酶也在这种方法中用于制备全长cDNA,制备好的cDNA可以选择使用PCR来进行扩增,随后在产物上加上接头,形成测序文库。直接cDNA测序会消除PCR偏倚,从而形成高质量的测序结果;但是,如果使用PCR来制备测序文库的话,需要的RNA数量更少。ONT cDNA测序法尚未报道过在PacBio测序仪上观察到的片段长度偏倚。
这两种长读长cDNA方法都受到标准模板切换逆转录酶使用的限制,这种逆转录酶能用全长RNA以及截短的RNA来生成cDNA。逆转录酶可以将那些只含5ʹ帽子结构的mRNA置换为cDNA,这样的话,那些由于RNA降解,RNA剪接或不完全cDNA合成而生成的短转录本就不会被反转录为cDNA,从而提高数据质量。但是,有报道指出,逆转录酶会对ONT平台的读长产生不良影响。
前面我们提到了长读长测序方法,这种测序方法与短读长测序平台一样,它们都依赖于将mRNA转换为cDNA。而最近Oxford Nanopore指出,他们的纳米孔测序技术可以直接对RNA进行测序,也就是说,这种测序手段不需要常规测建库过程中的的cDNA的合成和/或PCR扩增操作。这种方法称为dRNA-seq,这种方法就消除了常规建库过程中的偏倚,并且能够保留表观遗传学信息。这种方法可以从RNA直接进行两个接头的连接来制备文库。首先,带有一个oligo(dT)悬臂的双链核酸接头退火并连接到RNA的多聚腺苷酸(PolyA)尾部,随后就是可选(但不推荐的)的逆转录操作,这一步用于提高测序的通量。第二个连接操作就是添加测序接头,这个测序接头上已经提前安装有驱动测序的马达蛋白。文库随后进行MinION测序,其中RNA直接从3ʹpoly(A)尾部向5ʹcap端进行测序。最初的研究表明,dRNA-seq的测序长度过超过1000bp,最大测序长度过超过10kb。与短读长测序相比,这种长读长测序的几个优势在于:长读长测序可以提高对异构体的检测,并且它们还可以用于下方代码poly(A)尾巴的长度,这对于可变poly(A)分析( alternative poly(A) analysis)来说非常重要。Nanopolish-polya这个工具可以对那些用纳米孔测序得到的数据进行分析,计算出poly(A)尾的长度,这就包括基因之间的长度,也包括转录亚型之间的长度。这种分析证实了,保留内含子的转录本比完全剪接的转录本具有略长的poly(A)尾巴。虽然dRNA-seq还处于起步阶段,但是它具有检测RNA碱基修饰的潜力,因此它的应用潜力巨大,尤其是能够对表观遗传学转录进行新的分析。
虽然长读长技术在评估转录本方面比短读长技术有一些明显的优势,但是长读长技术也有一些明显的局限。尤其是与短读长技术相比,长读长技术的测序通量更低,错误率更多。但长读长技术的主要优势在于,它们能够捕获更多的单个转录本,不过这依赖于高质量的RNA文库。总体来说,这些局限影响了那些完全依赖于长读长测序实验的灵敏性(sensitivity)与特异性(specificity)。
长读长测序方法的主要局限就是当前的通量。在Illumina平台上,运行单次的RNA-seq可以生成10E9-10E10条短读长,但是在PacBio和ONT平台上,一次RNA-seq则只能产生10E6-10E7条读长。这种低通量限制了应用长读长测序技术进行实验的规模,并降低了对差异基因表达检测的灵敏性。然而,并非所有的实验都需要高深度测序。对于那些主要研究异构体的发现以及其特征的研究者们来说,测序长度比测序深度更重要。例如1百万个PacBio环形一致性测序(circular consensus-sequencing, CCS)的读长几乎就可以保证产生那些大于1kb的高表达基因的检测,ONT测序技术也是如此。因此,对于那些低到中等水平表达的基因来说,测序深度确实是一个主要问题。当进行同期功能基因组学分析(contemporary functional genomics analysis)大规模的DGE实验时,这种低通量测序技术的局限就会表现得明显。在这些研究中,必须对多个样本组进行分析,每组就是由多个生物学重复构成的,这样就能够实现充分的统计功效来有确认那些在整个转录组水平上发生的精确变化。对于这种需求,长读长技术不太可能取代短读长技术,除非长读长的测序读长的生成量能提高2个数量级。随着全长RNA-seq读长数目的增加,转录本检测的灵敏度将会增加到类似于Illumina平台上的这种水平,并同时具有更高的特异性。与此同时,通过将Illumina 的短读长RNA-Seq与PacBio的长读长Iso-Seq结合(并且可能还与ONT方法结合),可以增加全长RefSeq注释的异构体检测的数量、灵敏性和特异性,同时保留转录本量化的质量。虽然长读长RNA-seq方法目前的实验成本较高,但它们可以检测到短读长方法遗漏的异构体,尤其是那些难以测序但与临床相关的区域,例如高度多态的人类MHC或雄激素受体。
长读长测序平台的第二个主要局限就是其更高的错误率,它比成熟的Illumina测序仪要高出一到两个数量级。长读长测序平台上生成的数据还包含更多的插入-删除错误。虽然这些错误与识别变化(variant calling)有关,但在RNA-seq中,每个碱基都被正确识别并非那么重要而长读长测序的目标是要阐明转录本和异构体(While these error rates are of concern for variant calling, in RNA- seq it is less crucial that every base be called correctly, as the goal is only to disambiguate transcripts and isoforms)。这种错误率对于其应用来说确实是一个值得观注的问题,现在正在解决这一问题。PacBio SMRT测序平台上出现的随机错误通常可以通过使用CCS增加测序深度来进行解决,在这种技术里,cDNA经过长度选择和接头进行环化后,每个分子就可以被多次测序,从而产生长度范围是10-60kb的连续长读长,并且包含许多原始cDNA的拷贝。这些长读长经过数据分析后就被处理为单个cDNA子子读长(subreads),这些子读长被组合后就可以产生一致的序列。分子测序的次数越多,产生的错误率就越低;CCS已经被证明可以将错误率降低到与短读长相当的水平,甚至更低。但是,将更多的这个平台的测序能力用于重新读取相同的分子,就又加剧了其测序通量的问题,因为可以读取的唯一转录本变得更少了。
长读长RNA-seq方法的灵敏度还受到其他几个因素的限制。首先,它们依赖于长RNA分子以全长转录本的形式进行测序,但是要达到这种情况并非总能实现,因为在样品处理和RNA提取过程中RNA会发生降解或剪接。这种情况在短读长RNA-seq中也存在(3ʹ端的偏倚),但这种问题在短读长中是可控的,对于全长转录组分析进行研究的研究者们来说,即使是低水平的RNA降解,也能限制长读长的RNA-seq效果。因此,对于那些即将使用长读长进行测序的研究者来说,需要仔细地对提取的RNA进行质控。其次,中位数的读长长度会进一步受到文库制备中的技术问题与偏倚的限制,例如有些cDNA合成的截断或某些cDNA是由降解的mRNA合成的,最近开发的高效逆转录酶对此有所改进,这些酶有着更高的链特异性,甚至能够产生更多的3ʹ-5ʹ转录本的覆盖。虽然这些酶还未被广泛使用,但是这些高效逆转录酶也提高了结构稳定的RNAs,例如tRNAs的覆盖率,在oligo-dT和全转录组分析(WTA)方法中使用的逆转录酶很难处理这些结构稳定的RNAs。第三,长读长测序平台固有的偏倚(例如长文库分子在测序芯片表面上的低扩散)会降低更长转录本的覆盖率。
长读长方法(使用cDNA或dRNA-seq)解决了用于异构体分析的短读长测序方法中的一个基本问题,即它们的读长长度。长读长方法可以生成从Poly(A)尾部到5ʹcap的跨异构体的全长转录本读长。因此,这些方法使得分析转录本及其异构体成为可能,从而无需从短的读长中重构它们或推断它们的存在;每个测序的读长仅仅代表了它的起始RNA分子。全长cDNA测序或dRNA-seq用于分析DGE的未来应用将依赖于PacBio和ONT技术的更高通量。长读长RNA-seq分析正被研究者们迅速采用,并与深度短读长RNA-seq数据结合起来,用于更全面的分析,这非常类似于基因组组装所采取的混合方法。随着时间的推移,长读长和dRNA-seq方法可能会用于证明已经鉴定的基因和转录本的列表,即使在研究很透的生物中,对于基因和转录本的研究也还远远不够。随着方法的成熟,以及测序通量的增加,差异转录本分析将会成为常规方法。合成长读长RNA-seq或其它技术的发展将对这个领域产生什么样的影响,还有待观察。然而从目前来看,Illumina短读长RNA-seq依然占据了主导地位,在这篇综述的剩下部分中我们将会集中讨论短读长测序。

 首页
首页