RNA-seq的主要用途在于研究样本中的mRNA的种类与数量,但是mRNAs的存在与否并不直接关系到蛋白质的合成。现在有两种方法可以研究转录以外的翻译情况,可以让研究者们更好的理解翻译组(translatome):一种是多核糖体表达谱(polysomal profiling),一个是核糖体足迹RNA-seq(Ribo-seq)。核糖体对mRNAs的翻译具有高度的调节作用,蛋白质水平主要由翻译活性决定。多核糖体表达谱与Ribo-seq可以让研究者探索一个转录本占用多少个核糖体以及核糖体在转录本上的分布(FIG. 5)。这种方法可以让研究者推断在特定时间或细胞状态下哪些转录本正在被活跃地翻译。这两种方法都假设mRNA 核糖体的密度与蛋白质合成的水平相关。在不同样本之间进行比较,就能提示治疗条件下,时间推移以及疾病发展过程中,核糖体的动力学特征,上述的这些情况都与翻译的异常调控有关,例如纤维化,朊病毒或癌症。
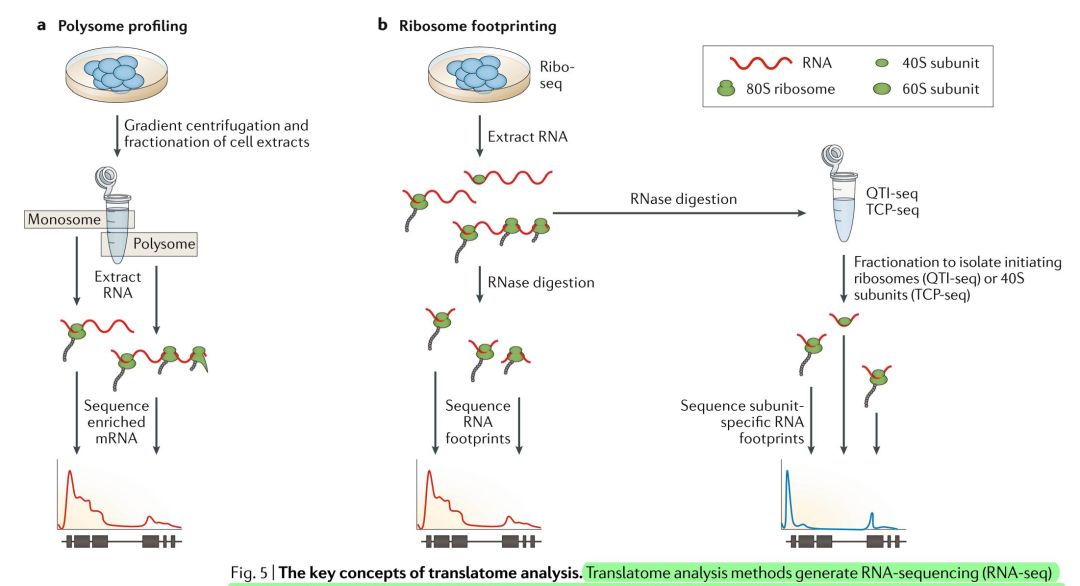
Figure 5-翻译组的关键概念。翻译组方法是从那些与核糖体结合的RNA中生成RNA-seq数据,这种方法假设mRNA上的核糖体的密度与蛋白质的合成水平相关。(a)多核糖体表达谱的方法是通过离心将RNA分子分成多核糖组分,然后通过RNA-seq的方法进行比较。在多核糖体组分中表达较高的RNA被认为是更活跃的转录。(b)核糖体足迹(Ribo-seq)法使用RNase来降解暴露的RNA,同时保留那些被核糖体保护的未被降解的RNA。通过对这些保护的RNA进行测序,就可以揭示出核糖体的密度与位置。通过修改变标准Ribo-seq方法,定量翻译起始测序(QTI-seq)或翻译复杂表达谱测序(TCP-seq)可以专门富集起始核糖体或其亚基,同时剔除延长的核糖体,因此可以对翻译的动态过程进行更详细的分析。对翻译组RNA-seq数据的过计算 分析可能确定每个mRAN的相对翻译程度,可以研究翻译的起始,延长与终止的动力学过程。
在多核糖体表达谱实验中,使用蔗糖梯度超离心将与多个核糖体(多核糖体组分)结合的mRNA和与单个核糖体结合的mRNA(单核糖体组分)分离开来,前者用于RNA seq文库制备(FIG. 5a)。与单核糖体组分中检测到的mRNA相比,在多核糖体组织中检测到的高丰度mRNAs可以被认为翻译得更频繁。这种方法也可以用于推测单个mRNAs的翻译状态,也可以用于生成高分辨率的核糖体占有信息与密度(尽管它无法确定核糖体的位置)。这类方法的原始方法已经进行了几项改进。例如,使用非线性蔗糖梯度改善了多核糖体收集,使多核糖体在不同浓度蔗糖溶液界面的收集过程更为简单,使用Smart-seq文库构建技术可以让研究者们分析仅10ng级的多核糖体mRNA,使用更高分辨率的蔗糖梯度和深度测序可以检测了转录本异构体的特异性翻译。然而,多核糖体表达谱实验生成的翻译组信息分辨率相对低,这一过程还比较费力,需要特殊的仪器,这就限制了其应用范围。
Ribo-seq是基于RNA足迹的方法,它最初用于酵母研究。这种方法用环己胺(cyclohexamide)来抑制翻译延伸,并诱导核糖体在mRNAs上停滞。用RNase I消化mRNA会留下20-30个核苷酸,这20-30个核苷酸就是受核糖体保护的足迹,这些足迹被处理后用于制备RNA-seq文库(FIG. 5b)。Ribo-seq能生成高分辨率的翻译谱,描绘核糖体丰度和单个转录本的位置。而多核糖体分析中无法提供核糖体的位置信息时,这说明有可能检测到了翻译的暂停,这些检查可以调节蛋白质的表达。当方法修改了缓冲液和对酶进行了优化后,就能更清楚地揭示Ribo-seq数据中3-bp的周期性,以及条形码和UMIs(检测单个分子的事件)。标准的RNA-seq工具可以用于Ribo-seq的计算分析,但最近已经出现了特定的工具用于寻找开放阅读框,用于差异或异构体水平的翻译分析,以及用于研究密码子偏倚。Ribo-seq的主要限制就是超速离心,以及由于核酸酶不同批次间的变化,以需要经验来确定RNase I的消化条件。
这些方法检测的是来自翻译起始、延伸和终止的信号的平均强度,但是对Ribo-seq的修改可使得其能够研究翻译动力学。定量翻译起始测序(Quantitative translation initiation sequencing, QTI-seq)通过化学“冷冻”和富集起始核糖体,同时从结合的mRNA中去除延长的核糖体来定位转录起始位点。翻译复杂谱测序(Translation complex profile sequencing, TCP-seq)也通过在组装成熟核糖体之前富集与40S核糖体小亚基结合的RNA来检测起始位点。然而,由于这种方法中保留了核糖体的完整性,也可以分析和比较80S核糖体组分,从而更全面检测翻译动力学(FIG. 5b)。
所有的翻译组方法在概念上都是相似的;它们假设mRNA核糖体的密度与蛋白质的合成水平相关。虽然它们的样本制备方案不同,但都需要大量的起始细胞数。最终,翻译组与RNA-seq结合起来研究基因的表达水平,并与蛋白质组学一道来研究蛋白水平,这可能就需要对mRNA的翻译进行一个广泛地理解。如果想要了解翻译组的更详细信息,可以阅读最近的综述。(比如我们生信技能树前面的推文)
Ribo-seq分析必看文献知识(四):核糖体与蛋白质合成相关生物知识
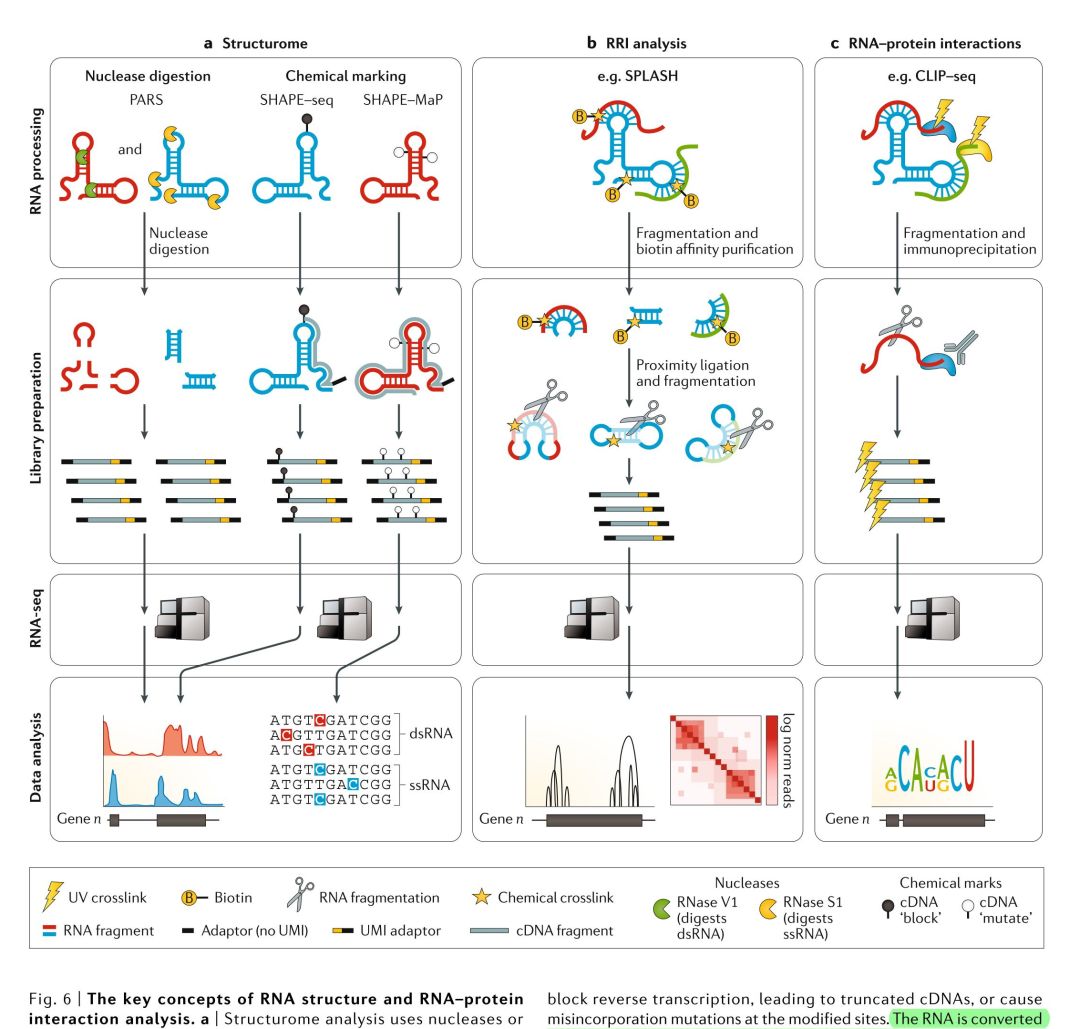
RNAs在调节其它生物分子和生物过程(例如剪接和翻译)中发挥着重要作用,它们涉及RNA与各种蛋白质和/或其它RNA分子的相互作用。RNA-seq可以用于研究分子内和分子间RNA-RNA的相互作用(RNA-RNA interactions, RRIs),这可能让研究者更好地理解结构组(structurome),或者是研究RNA与蛋白质之间的相互作用,这样就可以深入理解转录与翻译(FIG. 6)。针对相互作用组(interactome)分析而开发的各种方法都有一个共同的主题:在RNA中富集出那些与其它RNA有相互作用的RNA。一些方法利用的是天然生物学相互作用,而其它的方法则是在目标分子之间计算瞬时作用力或共价键;大多数方法使用的是抗体pull-dwon、亲和纯化或探针杂交的手段来富集RNA进行测序。在这里我们简要描述一下主要的基于RNA-seq的方法来研究结构组和相互作用体的内容。




 首页
首页


