

表1 梯度和pH的正交设计

表2 有关物质保留时间
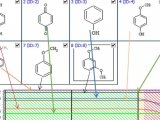
通过ACD软件构建梯度&pH两因素模型,模型如图1:

图1 梯度和pH的模型图,提示pH5.6范围下有全局最优


图2 pH5.6下的验证实验结果

表4 模型实测预测表

图3 去除辅料峰影响后的有关物质模型图

表5 pH 6.0下执行的梯度

图4 pH6.0下梯度表的实际数据图

各杂质分离良好,从模型上看,耐用性范围较宽,可作为最终检验方法进行预验证。
目前已经有了可用的方法,倒回来再研究pH5.6下的模型预测和实测失真的问题,在首次进行建模后验证时发现的pH5.6下葡萄糖加和物与杂质IV预测不准的问题。将此案例与ACD中国区经理的阎作伟沟通,被评价为“非常有意思的结果”,因为出现了主成分和加合物杂质在pH5和pH7两次靠近的现象。阎作伟经理表示这种情况可能发生,但实际上还是很少见。他提醒是否在建模时杂质定错了位。遂与实验实施人员复盘了所有数据,结果发现,杂质定位判断的确出错了,如图6a和图6b

图6a 错误的杂质定位判断

图6b 正确的杂质定位判断

图7 三个杂质的LogD叠加曲线

图8 定位修正后的模型能够准确预测出pH5.6的共流出
感谢天地恒一的案例,给读者带来了一个重要的提示。Peak Matching 一直是进行保留时间数学建模的难点之一。不论用自动化还是用手动Peak Matching都可以借助DAD,MS的结果做辅助。如果只是单波长下用色谱峰的形态进行Peak Matching,的确很考验眼力,如果是混标情况下,最好把加标的浓度配的不一样,这样也有利于提高辨识度。
by 阎作伟

















 首页
首页




