
本文内容是2022年3月-4月同写意系统化分析方法开发系列网络培训的第三节的部分内容。应听众的要求,我们将把此次系列演说的内容以文字的形式重现,以期令更多的读者可以有更深入的学习和讨论的机会。




今天给各位带来的内容分三部分。首先是和大家一起探讨我们常规的方法开发思路,其次通过一个起始物料的有关物质方法开发经历来阐述其中的科学思维,最后总结和分享了一些分析方法开发的经验供参考。

目前药物质量研究工作不仅限于原料药和制剂,关键起始物料的质量控制在整个项目申报中起着至关重要的作用,其研究内容之广不亚于一个原料药,包括有关物质、基因毒杂质、对映异构体、残留溶剂、无机杂质及含量等多项研究。而起始物料通常因为分子片段小、极性大、紫外吸收弱、活性大、异构体杂质多等特点,造成方法开发上保留弱、灵敏度差、不稳定及难分离等问题,所以需要结合理论知识,利用一些特殊色谱分离工具并综合考虑多方面的因素(包括色谱柱、流动相、检测器、前处理方式、稀释剂及稀释过程、进样量、检测波长、供试品浓度、定量计算方式及环境要求等)才能更好的开展这项工作。

1.明确试验目标,比如:
一个含量的方法,关注拖尾因子、理论塔板数
一个基因毒杂质的方法,最关注的是灵敏度;
一个包含多个杂质的有关物质方法,需要关注的是分离度;
2.对目标物进行理论分析和风险评估
(ACD公司Percepta软件可以预测化合物的物理性质,包括溶解性、logP/logD、pKa等)

首先了解化合物的logD值:logD值大小代表着化合物疏水性强弱,进而影响化合物在反相色谱上的保留,即logD值越大,保留越强;logD值越小,保留越弱;故logD值对色谱柱填料的选择非常重要。当logD值大于0,可以选择普通C18;当logD值小于0,可以选择AQ、RP色谱柱等,保证极性化合物有一定的保留;当logD值小于负3,可以选择离子对色谱,此时离子对试剂的浓度和缓冲液的pH就是关键参数。
其次要了解化合物的pKa:pKa值显示化合物的一种离子化能力,当pH=pKa时,溶液中离子化的分子与中性分子浓度相同;而我们又知道,化合物电离程度越高,logD越小,在反相色谱上的保留越弱;所以pKa值会影响我们选择流动相的pH值。我们要尽量避开pKa值±1范围,特别是主成分的pKa值,保证主峰峰型良好以及方法的耐用性。
然后要关注化合物结构上的一些特殊基团:如果化合物中含有活泼羰基或醛基,那么需要避免使用甲醇作为流动相和溶剂;如果化合物中含有F、OH、醚基、酰胺等氢键作用位点的基团,可以优先选择甲醇作为流动相;如果化合物中含有硝基、苯环上取代异构,可以对比甲醇和乙腈或二者混合比例的选择性差异。如果化合物需要含有(CC双键、羰基、醛基、偶氮基、亚硝基等)发色基团时,我们就可以选择紫外检测器;如果化合物中没有这些基团(比如:糖类),我们需要选择通用型检测器。
最后还需要关注化合物的溶解性:溶解性对选择溶剂和样品是否需要特殊的预处理有指导意义;溶剂选择的不恰当,会产生溶剂化效应,导致峰型差或双峰等问题。

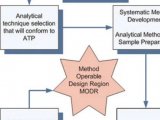
3. 基于上述的理论知识分析和风险评估,可以锁定一个初始的色谱条件。
4. 找关键参数:对于简单的方法,通过几组试验就可以找到关键参数;但如果面临的是比较复杂的体系,矛盾点较多,人脑不够用的时候,可以借助多功能方法开发系统同时筛选色谱柱、流动相A的pH值、有机相种类、梯度、温度等,用最短的时间找出关键参数。
5. 在小范围内优化关键因素指标。
6. 探索最佳耐用性空间:可以借助ACD计算机辅助软件探索最佳耐用性空间,以保证我们方法在验证、转移、甚至整个生命周期都是可以适用的。
7. 制定合理的控制策略:主峰与相邻杂质杂质峰分离度,增加关键分离对的Rs等。

本案例分离目标是主成分(ID6)及7个相关杂质,在开发之前先进行结构分析:
从logD曲线叠图可知:这组化合物在pH1~9范围内logD均大于0,所以保留不是问题,在设置梯度时起始有机相比例可适当提高;但同时ID5和ID8、ID6和ID7几乎重叠,说明想完全通过疏水性的差异来分离的可能性比较小;观察ID5和ID8结构,主要差异是甲氧基在苯环上的取代位置不一样,甲氧基作为供电子基团,在苯环上邻位/间位取代异构,会导致苯环上电子云密度分布差异;同时观察到ID6和ID7也是苯环上相差一个甲氧基的对位取代,同样会造成苯环上的电子云密度分布差异,可以利用苯环上π-π相互作用差异来进行分离,优先考虑苯基柱系列的色谱柱(Phenyl、PFP或C18-PFP等)。

各化合物除了ID1、ID3、ID4有弱酸基团酚羟基外,其他化合物均无酸碱基团,酚羟基pKa在10左右,而我们在建立方法的时候,通常不会考虑pH大于11以上的方法,所以本方法pH不会是关键参数。

最后我们再来看化合物上的特殊基团,这组化合物都含有苯环,可以选择紫外检测器;含有羟基、醚键、羰基等氢键作用位点,可以优先选择质子供体甲醇作为流动相,前面分析过该组化合物在苯环上的取代基不同会造成苯环上的电子云密度分布差异,乙腈作为偶极溶剂能在色谱柱及化合物之间产生或抑制π-π键的作用,会产生不一样的选择性差异,所以甲醇和乙腈的混合比例是关键参数。

在方法摸索过程中分离度Rs是我们主要关注的目标函数,我们需要根据分离度经验公式不断调整优化策略,公式如下:

其中α与Rs是一次线性的关系,所以α对分离度值是影响最大的参数,也是我们分离过程中需要关注的主要矛盾。


根据理论分析,本案例甲醇的选择性可能会比乙腈更好,故首先考察了流动相B为100%甲醇时的分离情况,结果经过优化后,除关键分离对ID6和ID7以外(ATP),其他杂质的分离和保留均较好。

为进一步改善ID6和ID7的分离情况(优化选择性α),考虑在100%甲醇中加入不同比例的乙腈对比选择性差异,结果如图所示,当乙腈含量较高时,ID6比ID7先洗脱;logα<0(k1为ID6,k2为ID7),当乙腈与甲醇比例1:1时,logα=0(k1=k2),ID6与ID7共洗脱;当乙腈含量较低时,ID6比ID7后洗脱,logα>0。

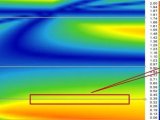
以甲醇在乙腈中的比例作为横坐标,logα作为纵坐标作图,可以看出当点穿过横坐标时(logα=0)分别位于第I和第IV象限,洗脱顺序出现了翻转,90%的甲醇-乙腈的分离最好。但为了保证方法的耐用性,我们需要找到最佳的可操作空间,故结合ACD软件分别进行了二维、三维模拟实验。

根据文献和经验可知,柱温和梯度对结构类似物的分离很重要,故先设计了温度(logk=logA+B/Tk)与梯度(logk=logkw-φS)的2因素2*3的实验。利用ACD/LC-Simulator创建梯度温度2D模型,通过模拟结果发现,ID6与ID7在梯度时间较短的范围内可以基线分离的空间较窄,方法耐用性可能不好,故需继续设计三维模拟实验。

根据前面研究的结果,明确了分离目标,构建了一定的知识空间,也通过方法筛选对关键的参数进行了简单的风险评估,最后再针对性的设计了包含梯度、混合有机相比例及温度3因素的DOE试验,并构建3D模型。通过对模型的分析,柱温越低分离越好,梯度时间越长分离也越好,关键分离对ID6与ID7在高比例甲醇或者乙腈下都可以找到一个合适的方法,但是当乙腈含量较高时,ID7在ID6(主成分)之后出峰,在不同品牌仪器上进行方法转移或柱效下降时,分离的风险较大。


最后根据三维模拟的结果选择最佳的色谱条件为:有机相甲醇-乙腈的比例为9:1;柱温25℃;线性梯度时间为35分钟,其他与初始色谱条件保持一致。ACD软件预测的色谱图与实际测定的色谱图基本一致,关键分离对的Rs达到3.0以上。

前面讲到苯基柱系列的色谱柱对苯环上取代基位置异构体杂质选择性良好,那普通C18色谱柱是否可以呢?这一类异构体杂质因为疏水性和酸碱性几乎一致,从理论分析上苯基柱系列是最合适的,前面我们先入为主的选择了C18-PFP,试验结果也是很满意。但我们应该还可以从氢键作用、π-π与空间位阻等作用力等来继续考察其他色谱柱的分离情况。

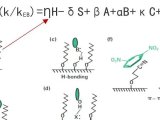
经典的施耐德疏水减法模型,只适用于苯基柱和氰基柱以外的色谱柱,也就是等式1:这里的大写字母表述的是色谱柱的参数(H:疏水性、S*:位阻、A:氢键酸度、B:氢键碱度、C:离子交换活度),希腊字母(η’、σ‘、β‘、α‘、κ‘)是相对应的化合物性质;异构体杂质大部分logD在任何pH条件下都几乎重合(疏水性和酸碱性几乎无差别),怎样才能分离它们呢?只有利用填料结构差异,氢键作用与π-π作用等。我们都知道在苯基柱和氰基柱上存在着π-π或偶极-偶极相互作用,等式1并不适用。随着色谱理论的发展,2005年分析科学家在等式1的基础上对π-π或偶极-偶极相互作用进行了概念上的补充,即等式2,描述了色谱柱与化合物之间的6大作用力(分别是疏水相互作用、空间位阻、氢键作用、离子交换、π-π作用、偶极-偶极相互作用),从而帮助我们更好的理解反相色谱保留机制,也为我们如何选择合适的色谱柱奠定了基础。
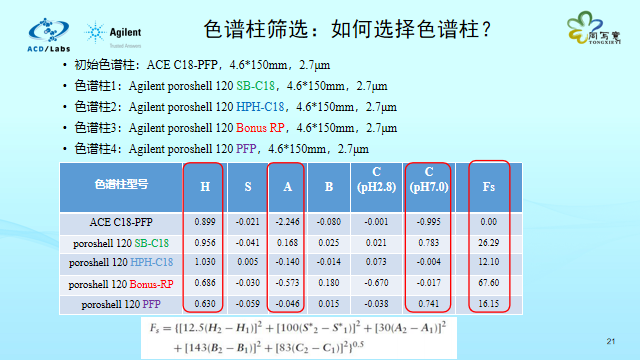
在方法开发初期阶段,面对形形色色色的谱柱,如何挑选合适的色谱柱进行筛选?为了评价两根色谱柱的相似度,分析科学家引入了色谱柱-比较函数Fs,分别从色谱柱的5大参数进行比较。如果Fs值小于3,说明两根色谱柱选择性几乎一致;如果Fs值大于50,说明两根色谱柱选择性相差非常大。在方法开发初期,我们尽量选择Fs值相差较大的几根色谱柱来筛选,以提高选择性差异。我们选择了4根色谱柱,通过查询这5根色谱柱的参数,由于疏水性H、氢键酸度A、pH=7.0下的离子交换值C的差异,导致Fs值相差很大,最大程度地提高筛选的范围。

在方法开发阶段,可以使用Agilent多功能方法开发系统和ACD在线联用,快速筛选的同时数据也可以直接导入ACD软件进行数据建模。

在乙腈体系下,ID1和ID2在SB-C18和HPH-C18分离良好,在Bonus RP和PFP上共洗脱;另两组关键分离对ID3和ID4、ID6和ID7在四根色谱柱上分离均良好。总体而言,四根色谱柱上选择性差异较大。

在甲醇体系下,ID1和ID2在Bonus RP和PFP上分离明显改善,达到基线分离;ID3和ID4在四根色谱柱上分离均良好,但是洗脱顺序发生翻转;ID6和ID7在SB-C18上分离度变差,其他三根色谱柱上分离良好。

为什么甲醇和乙腈的选择性不一样呢?回归到甲醇、乙腈结构上来分析。
乙腈作为偶极溶剂,可以与化合物之间发生偶极-偶极作用;π-π相互作用可以发生在任何不饱和的化合物之间;比如图片中,1,3-二硝基苯,吸电子的硝基取代芳香烃,是π-酸性分子,可以与π-碱性(比如以苯分子为代表的)之间发生π-π相互作用,乙腈作为一种π-酸性分子,可以与芳香类化合物分子之间发生π-π相互作用;同时乙腈还可以抑制化合物与色谱柱之间的π-π相互作用。甲醇作为酸性(质子供体)溶剂,可以与一种碱性分子(N,N-二甲基苯胺)N原子上的孤对电子相互作用,形成氢键;四氢呋喃作为碱性分子(质子受体)可以与苯酚上的H原子相互作用,形成氢键。

针对ID3和ID4在甲醇和乙腈体系下洗脱顺序翻转问题做了进详细的研究。用HPH-C18,研究不同比例甲醇-乙腈下分离情况:在纯乙腈体系中,ID4比ID3先洗脱;在甲醇-乙腈(50:50)体系中,ID4和ID3发生共洗脱;在纯甲醇体系中,ID3比ID4先洗脱。


为了更深入的理解ID3和ID4在不同甲醇-乙腈比例下的洗脱情况,我们设计了更多的实验点,并进行了数学分析,以甲醇-乙腈比例做横坐标,以logk做纵坐标,进行线性回归分析发现:二次项比一次线性回归分析更好。

混合有机相比例到底是一次线性关系还是二次项关系呢?化合物的保留与有机相比例的经验公式logk=logkw-φS,对于规则样品,是一次线性关系;而对于非规则样品,通常会偏离一次线性关系的,二次项拟合就有一定的实际意义。本案例中,甲醇-乙腈不仅有洗脱能力的差异,还有选择性差异,因此会偏离一次线性关系,二次项拟合更加准确。

ACD/Autochrom里内置公式,梯度拟合是一次线性拟合函数;混合有机相比例拟合是包含二次项的函数,由此可见ACD/Autochrom软件模拟准确度是理论支持的,多个项目经验也证实了这一点。

由化合物的保留与有机相比例的经验公式logk=logkw-φS,可以推导出两个化合物的的选择性与有机相比例的公式logα=(logkw3-logkw4)+φ(S4-S3),以甲醇-乙腈比例做横坐标,以logα做纵坐标,进行线性回归分析同样可以发现:二次项比一次线性回归分析更好,再次证明了甲醇和乙腈选择性差异。

利用ACD软件对确定的方法做了详细的耐用性考察,结果色谱条件在一定范围内进行变动时,各杂质以及关键分离对ID6和ID7的分离度均能符合要求(大于2.0),方法耐用性良好。

最后我们一起再来总结一下方法开发流程:
1. 铭记分离度基本公式
从保留因子k、选择性a、柱效N三个方面来改善分离度,选择性改变是就最有效的;
2. 明确开发目标
RS、TF/As、N、灵敏度;
3. 了解常用色谱柱的填料结构:
比如普通C18,有封端良好的,抑制碱性化合物拖尾;有不封端的,部分裸漏的硅醇基选择性较好;
比如镶嵌型的RP色谱柱,在C18链上镶嵌了极性酰胺基团,再比如AQ色谱柱,在硅醇基上键合亲水基团,对亲水化合物选择性和保留均较好;
比如苯基柱系列,苯基己基柱和五氟苯基柱,分别是富电子和缺电子的,选择性差异不一样,对苯环上的不同取代基、位置异构体有很好的选择性;还有联苯柱,空间位阻较大,对蛋白质构型差异也有识别能力。
比如氰基柱,含有-CN、-NO3基团的化合物选择性较好;
比如氨基柱,含有-NH2或其他极性的化合物选择性和保留较好;
比如其他新型混合型色谱柱,同时保留了C18的疏水性、镶嵌了极性基团、阴阳离子交换基团,除了对常规的疏水性化合物有保留外,还对一些钠离子、钾离子、氯离子、碘离子、硫酸根、柠檬酸根等都有所保留。
4. 了解常见化合物酸碱基团的pKa值:
羧酸根,pKa在3-5之间;
脂肪胺,pKa在7~11之间;芳香胺,pKa在3~7之间;
化合物的pKa值会影响选择缓冲盐pH值,尽量避开pKa±1;但是在选择缓冲对时又要尽量选择pKa±1:
常用的磷酸有三个pKa,分别是2.1、7.2、12.3,有三个缓冲范围,分别是1.5~3.5,6.0~8.5,11.0~13.5;
甲酸的pKa是3.8,缓冲范围就是2.5~5.0之间;乙酸的pKa是4.8,缓冲范围就是3.5~6.0之间;氨水的pKa是9.2,缓冲范围就是8.0~10.5之间;
在建立方法的时候,这几组缓冲盐可以满足大部分试验。但使用的时候需要注意:磷酸盐是不挥发的,在杂质谱研究的时候(LCMS),需要用甲酸或者甲酸胺或乙酸铵或氨水等替换。
5. 熟记常见的作用力类型
疏水相互作用、空间位阻、氢键作用、偶极-偶极作用、π-π相互作用、离子交换;
某些分析物与金属离子之间会发生螯合作用,在反相色谱上会带来一些负面影响(峰拖尾、保留时间漂移等),通常在流动相中加入EDTA或亚甲基二磷酸就可以解决(但还要考虑不同pH的络合能力差异);
常用的溶剂(甲醇是质子供体、四氢呋喃是质子受体,可以形成氢键作用;乙腈作为偶极溶剂,可以发生偶极-偶极作用;同时作为π-酸性分子,可以与化合物发生π-π相互作用,抑制化合物与色谱柱的π-π相互作用)。
6.先抓主要矛盾、再调整次要矛盾
先筛选带有两个+号的色谱条件(色谱柱类型、有机相类型、缓冲液pH值、离子对浓度、有机相比例),然后优化流速、柱温与进样体积等参数。
7.利用多功能筛选系统进行筛选和开发
从硬件上解放实验员;同时可以借助minitab、JMP等统计学软件,帮助我们设计试验和统计分析;利用ACD软件对试验数据建模,构建知识空间和探索最佳耐用性空间。
8.制定最终的控制策略
对方法学进行验证,检测代表性批次样品,确定关键分离度的Rs和灵敏度等定入质量标准,来保证方法的系统适用性。
杂质谱研究不仅仅是原料药和制剂成品关注的重点,也是起始物料最关注和最难的点,我们通常采取分步控制的策略,从源头控制,这样可以降低最终成品质量研究的压力和成本。不同的项目合成路线不同,化合物结构千差万别,杂质谱研究研无止境,随着新药研究的逐步发展,对于分析研发人员的方法开发能力越来越挑战,但无论什么项目我们都应该从化学结构的根本出发,采用科学的思维方法,理解被分离物质的特性并结合色谱理论进行分析,同时借助一些特殊的色谱工具(如日新月异的色谱柱填料)、多功能方法开发系统、ACD计算机辅助软件等,告别传统的试错”研究模式,一定能给我们的质量研究工作带来事半功倍的效果。

点击"阅读原文",查看完整视频














 首页
首页




